

A recurrent de novo splice site variant involving DNM1 exon 10a causes developmental and epileptic encephalopathy through a dominant-negative mechanism
- PMID: 36413998
- PMCID: PMC9748255
- DOI: 10.1016/j.ajhg.2022.11.002
A recurrent de novo splice site variant involving DNM1 exon 10a causes developmental and epileptic encephalopathy through a dominant-negative mechanism
Abstract
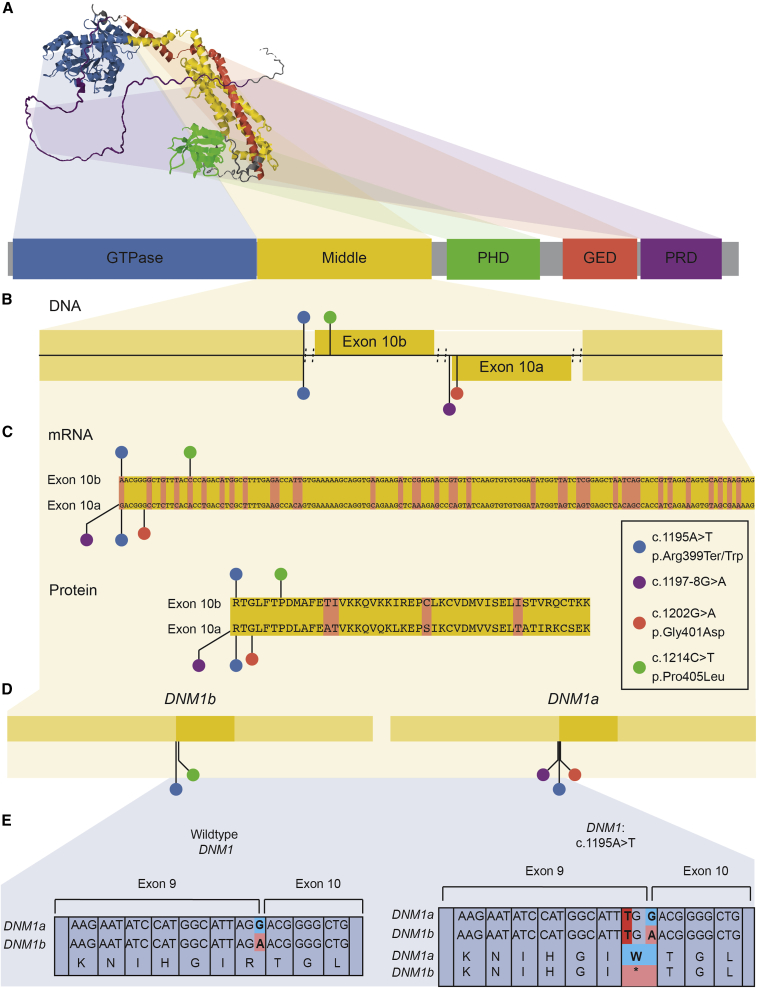
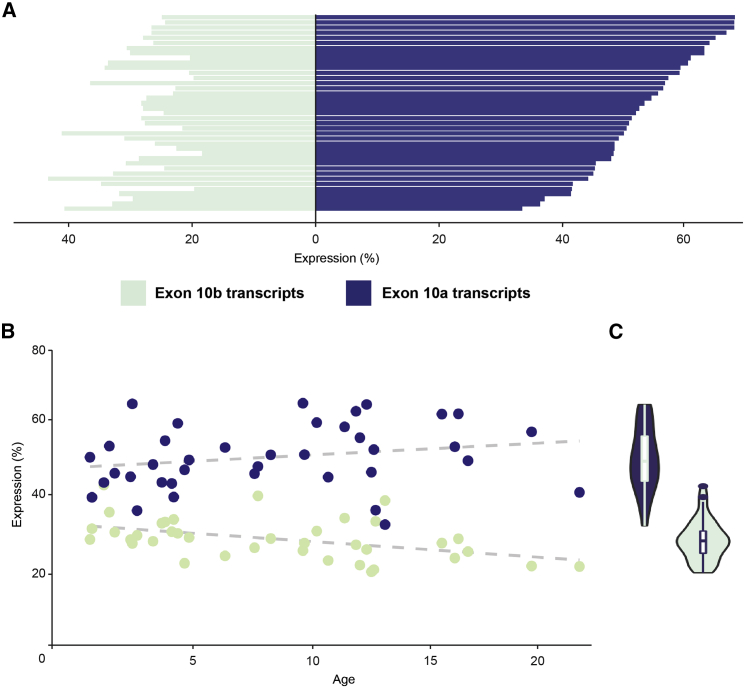
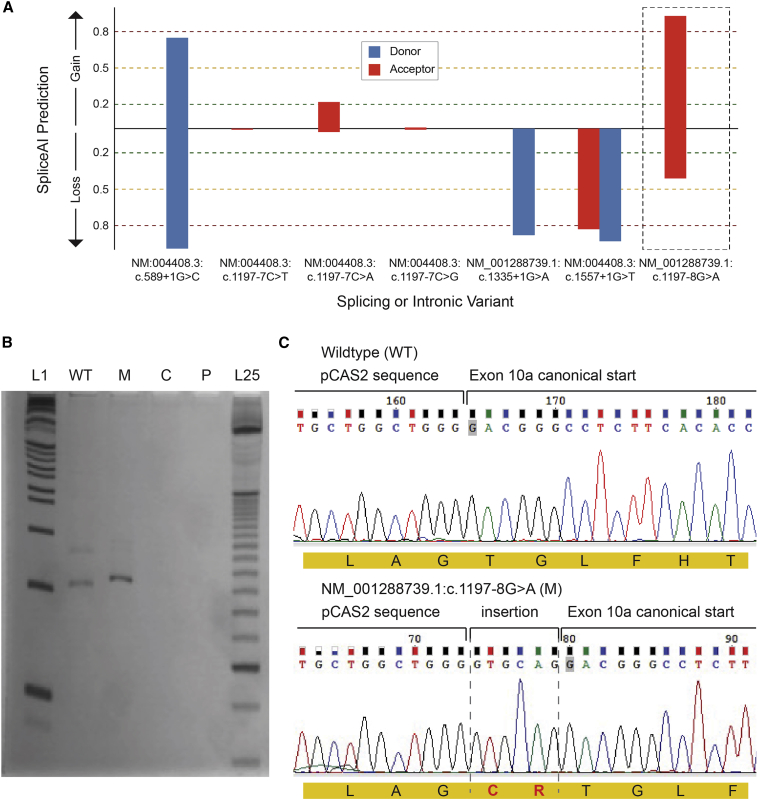
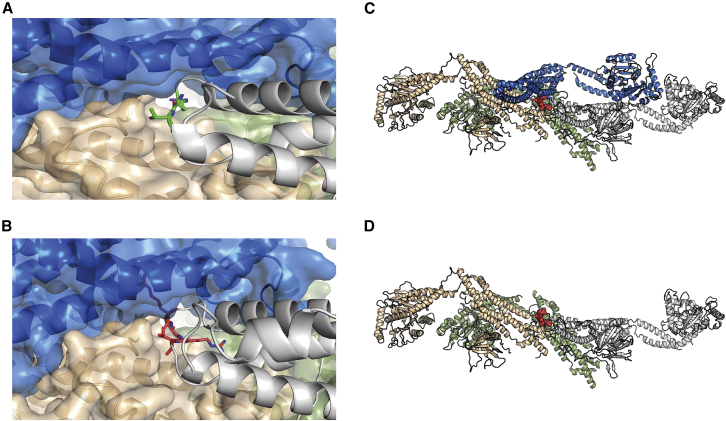
Heterozygous pathogenic variants in DNM1 cause developmental and epileptic encephalopathy (DEE) as a result of a dominant-negative mechanism impeding vesicular fission. Thus far, pathogenic variants in DNM1 have been studied with a canonical transcript that includes the alternatively spliced exon 10b. However, after performing RNA sequencing in 39 pediatric brain samples, we find the primary transcript expressed in the brain includes the downstream exon 10a instead. Using this information, we evaluated genotype-phenotype correlations of variants affecting exon 10a and identified a cohort of eleven previously unreported individuals. Eight individuals harbor a recurrent de novo splice site variant, c.1197-8G>A (GenBank: NM_001288739.1), which affects exon 10a and leads to DEE consistent with the classical DNM1 phenotype. We find this splice site variant leads to disease through an unexpected dominant-negative mechanism. Functional testing reveals an in-frame upstream splice acceptor causing insertion of two amino acids predicted to impair oligomerization-dependent activity. This is supported by neuropathological samples showing accumulation of enlarged synaptic vesicles adherent to the plasma membrane consistent with impaired vesicular fission. Two additional individuals with missense variants affecting exon 10a, p.Arg399Trp and p.Gly401Asp, had a similar DEE phenotype. In contrast, one individual with a missense variant affecting exon 10b, p.Pro405Leu, which is less expressed in the brain, had a correspondingly less severe presentation. Thus, we implicate variants affecting exon 10a as causing the severe DEE typically associated with DNM1-related disorders. We highlight the importance of considering relevant isoforms for disease-causing variants as well as the possibility of splice site variants acting through a dominant-negative mechanism.
Keywords: DNM1; alternative transcripts; developmental and epileptic encephalopathy; dominant negative; dynamin-1; epilepsy; synapse; vesicle fission.
Copyright © 2022 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
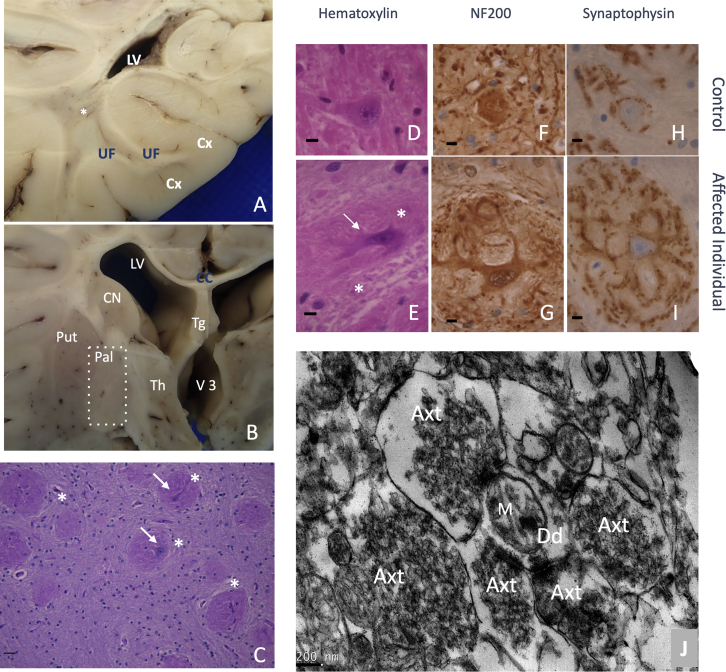
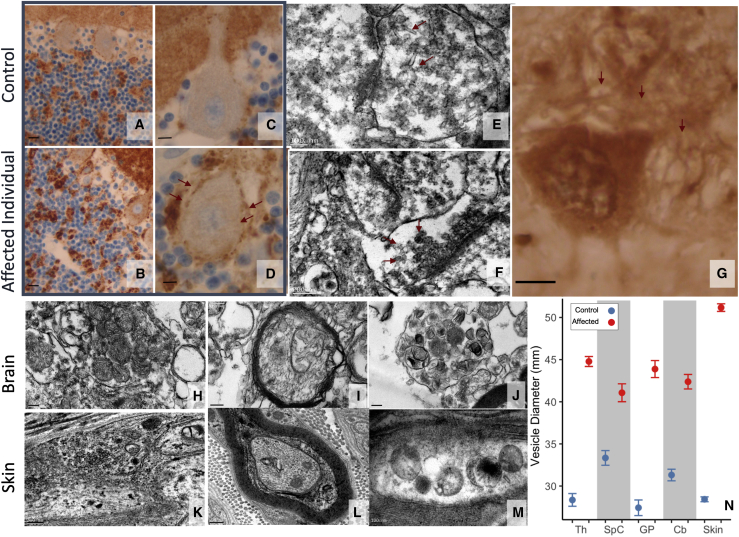
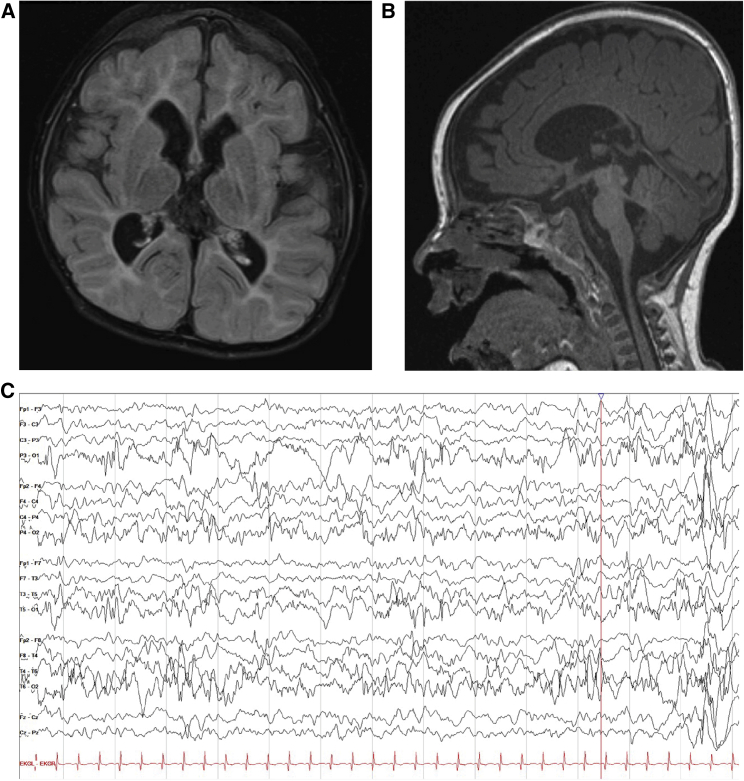
References
-
- Helbig I., Heinzen E.L., Mefford H.C., International League Against Epilepsy Genetics Commission Genetic literacy series: Primer part 2-Paradigm shifts in epilepsy genetics. Epilepsia. 2018;59:1138–1147. - PubMed
-
- McTague A., Howell K.B., Cross J.H., Kurian M.A., Scheffer I.E. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016;15:304–316. - PubMed
-
- Helbig K.L., Farwell Hagman K.D., Shinde D.N., Mroske C., Powis Z., Li S., Tang S., Helbig I. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet. Med. 2016;18:898–905. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
