

Widespread hypertranscription in aggressive human cancers
- PMID: 36417526
- PMCID: PMC9683723
- DOI: 10.1126/sciadv.abn0238
Widespread hypertranscription in aggressive human cancers
Abstract
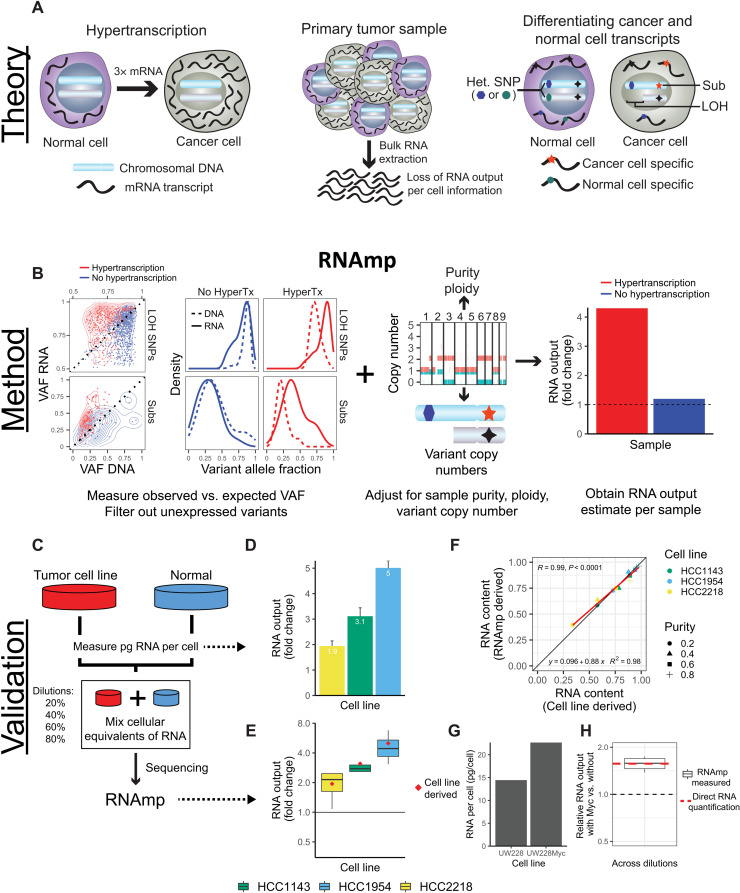
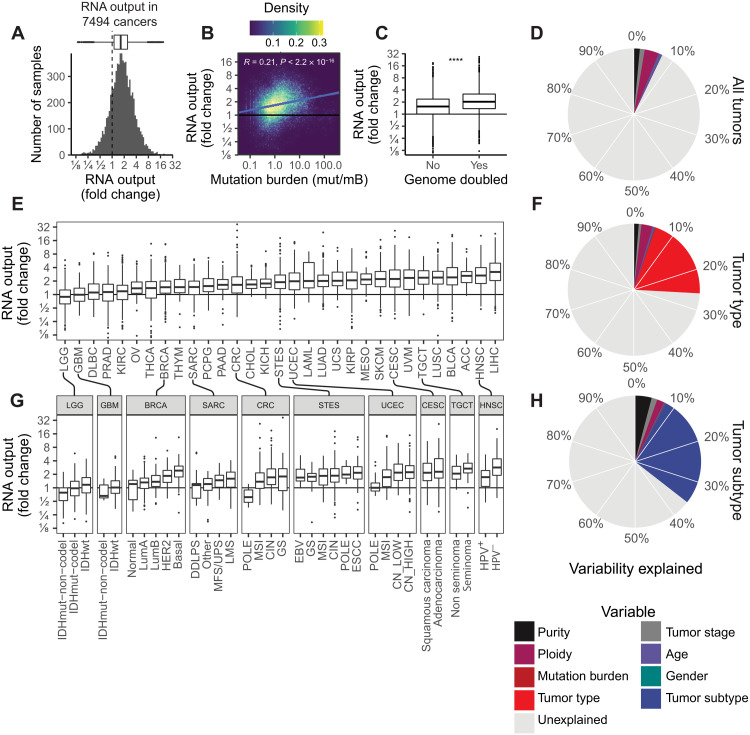
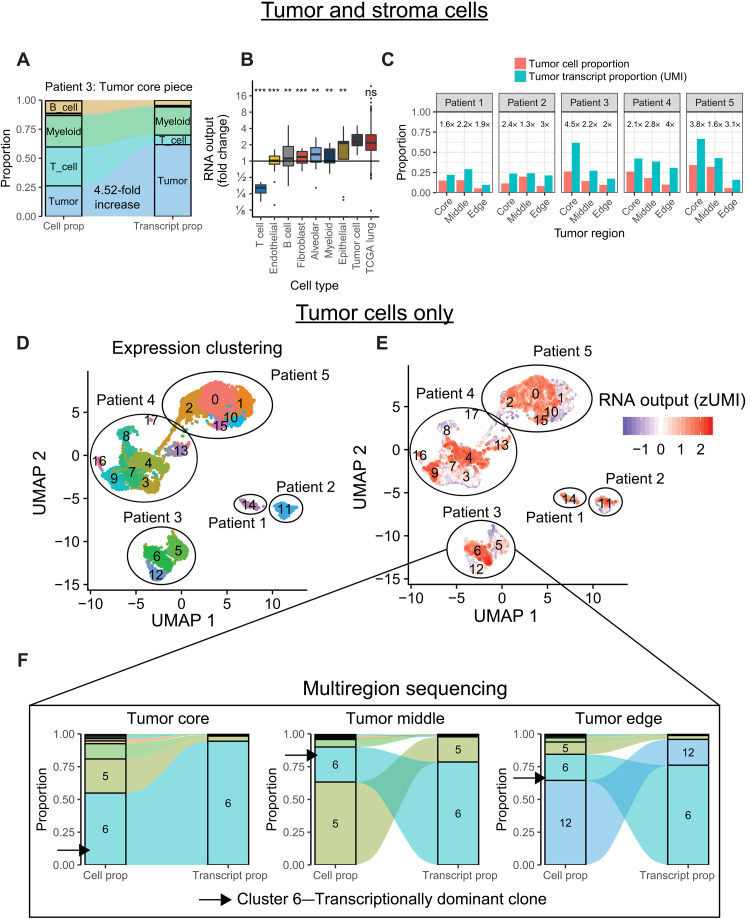
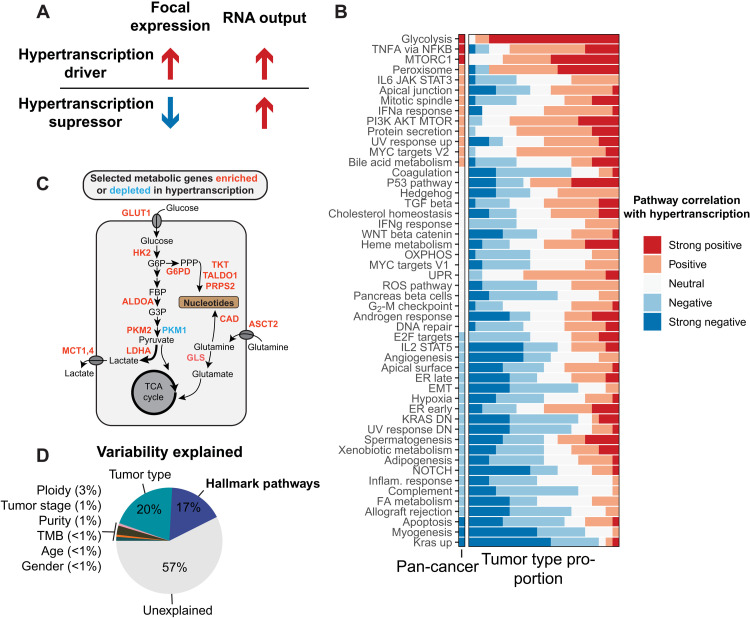
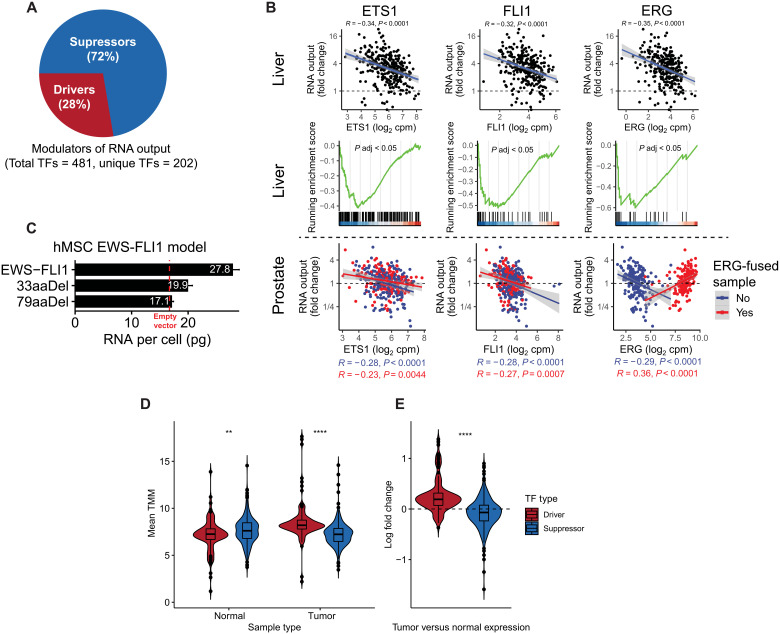
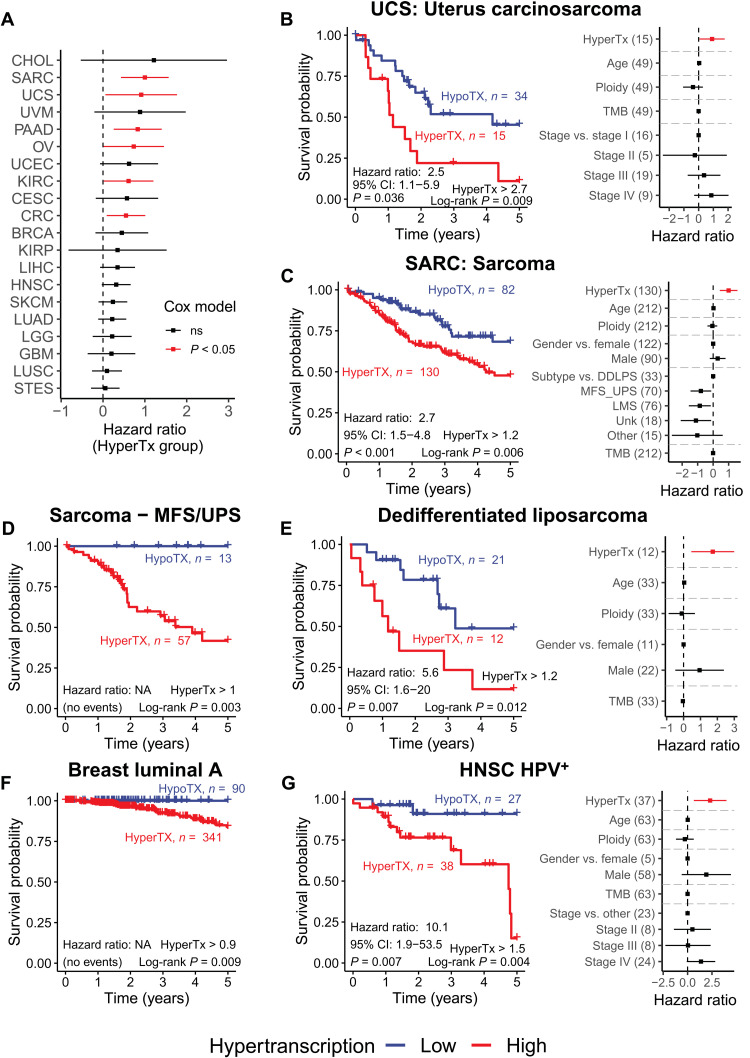
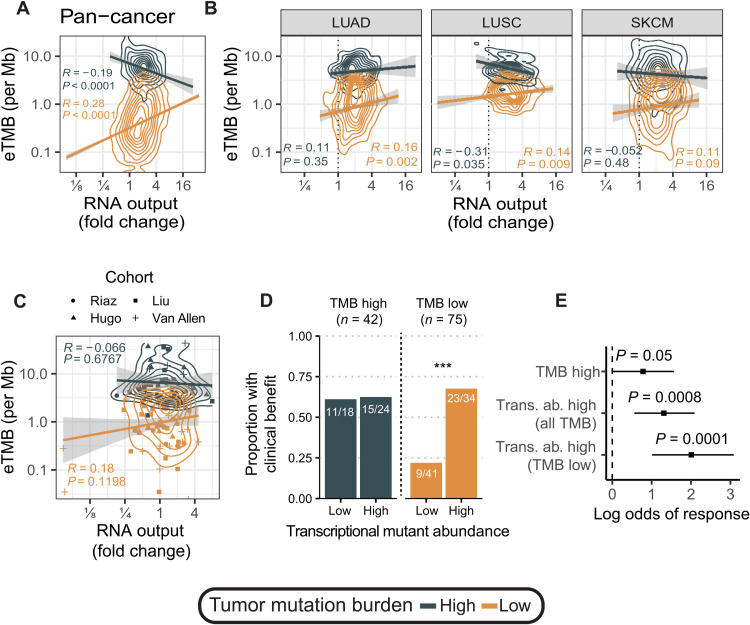
Cancers are often defined by the dysregulation of specific transcriptional programs; however, the importance of global transcriptional changes is less understood. Hypertranscription is the genome-wide increase in RNA output. Hypertranscription's prevalence, underlying drivers, and prognostic significance are undefined in primary human cancer. This is due, in part, to limitations of expression profiling methods, which assume equal RNA output between samples. Here, we developed a computational method to directly measure hypertranscription in 7494 human tumors, spanning 31 cancer types. Hypertranscription is ubiquitous across cancer, especially in aggressive disease. It defines patient subgroups with worse survival, even within well-established subtypes. Our data suggest that loss of transcriptional suppression underpins the hypertranscriptional phenotype. Single-cell analysis reveals hypertranscriptional clones, which dominate transcript production regardless of their size. Last, patients with hypertranscribed mutations have improved response to immune checkpoint therapy. Our results provide fundamental insights into gene dysregulation across human cancers and may prove useful in identifying patients who would benefit from novel therapies.
Figures
References
-
- Bielski C. M., Zehir A., Penson A. V., Donoghue M. T. A., Chatila W., Armenia J., Chang M. T., Schram A. M., Jonsson P., Bandlamudi C., Razavi P., Iyer G., Robson M. E., Stadler Z. K., Schultz N., Baselga J., Solit D. B., Hyman D. M., Berger M. F., Taylor B. S., Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 50, 1189–1195 (2018). - PMC - PubMed
-
- Li Y., Roberts N. D., Wala J. A., Shapira O., Schumacher S. E., Kumar K., Khurana E., Waszak S., Korbel J. O., Haber J. E., Imielinski M.; PCAWG Structural Variation Working Group, Weischenfeldt J., Beroukhim R., Campbell P. J.; PCAWG Consortium , Patterns of somatic structural variation in human cancer genomes. Nature 578, 112–121 (2020). - PMC - PubMed
-
- Nik-Zainal S., Alexandrov L. B., Wedge D. C., Van Loo P., Greenman C. D., Raine K., Jones D., Hinton J., Marshall J., Stebbings L. A., Menzies A., Martin S., Leung K., Chen L., Leroy C., Ramakrishna M., Rance R., Lau K. W., Mudie L. J., Varela I., McBride D. J., Bignell G. R., Cooke S. L., Shlien A., Gamble J., Whitmore I., Maddison M., Tarpey P. S., Davies H. R., Papaemmanuil E., Stephens P. J., McLaren S., Butler A. P., Teague J. W., Jönsson G., Garber J. E., Silver D., Miron P., Fatima A., Boyault S., Langerod A., Tutt A., Martens J. W. M., Aparicio S. A. J. R., Borg Å., Salomon A. V., Thomas G., Borresen-Dale A. L., Richardson A. L., Neuberger M. S., Futreal P. A., Campbell P. J., Stratton M. R., Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993 (2012). - PMC - PubMed
-
- Campbell B. B., Light N., Fabrizio D., Zatzman M., Fuligni F., de Borja R., Davidson S., Edwards M., Elvin J. A., Hodel K. P., Zahurancik W. J., Suo Z., Lipman T., Wimmer K., Kratz C. P., Bowers D. C., Laetsch T. W., Dunn G. P., Johanns T. M., Grimmer M. R., Smirnov I. V., Larouche V., Samuel D., Bronsema A., Osborn M., Stearns D., Raman P., Cole K. A., Storm P. B., Yalon M., Opocher E., Mason G., Thomas G. A., Sabel M., George B., Ziegler D. S., Lindhorst S., Issai V. M., Constantini S., Toledano H., Elhasid R., Farah R., Dvir R., Dirks P., Huang A., Galati M. A., Chung J., Ramaswamy V., Irwin M. S., Aronson M., Durno C., Taylor M. D., Rechavi G., Maris J. M., Bouffet E., Hawkins C., Costello J. F., Meyn M. S., Pursell Z. F., Malkin D., Tabori U., Shlien A., Comprehensive analysis of hypermutation in human cancer. Cell 171, 1042–1056.e10 (2017). - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
