

Dynamic quality control machinery that operates across compartmental borders mediates the degradation of mammalian nuclear membrane proteins
- PMID: 36417855
- PMCID: PMC9827541
- DOI: 10.1016/j.celrep.2022.111675
Dynamic quality control machinery that operates across compartmental borders mediates the degradation of mammalian nuclear membrane proteins
Abstract
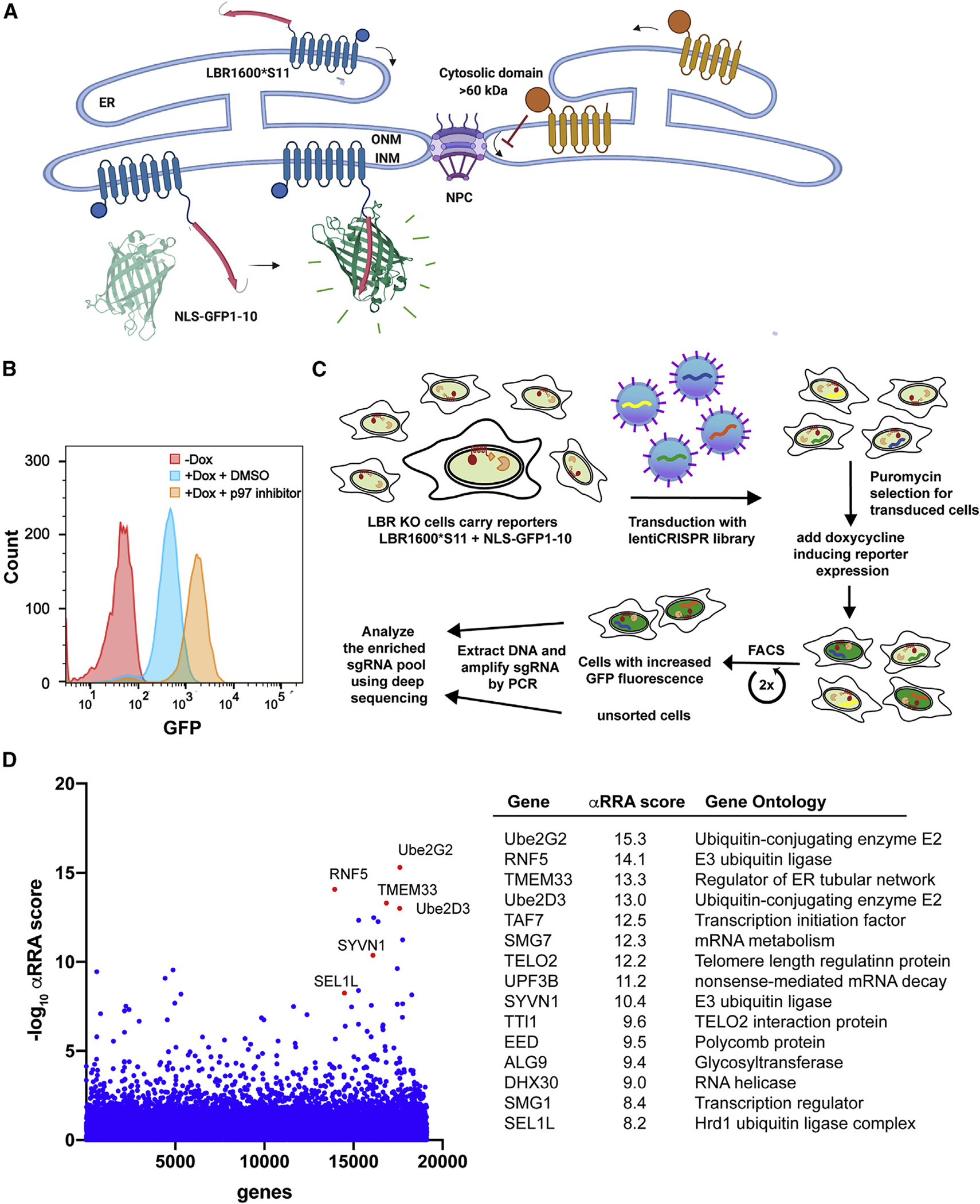
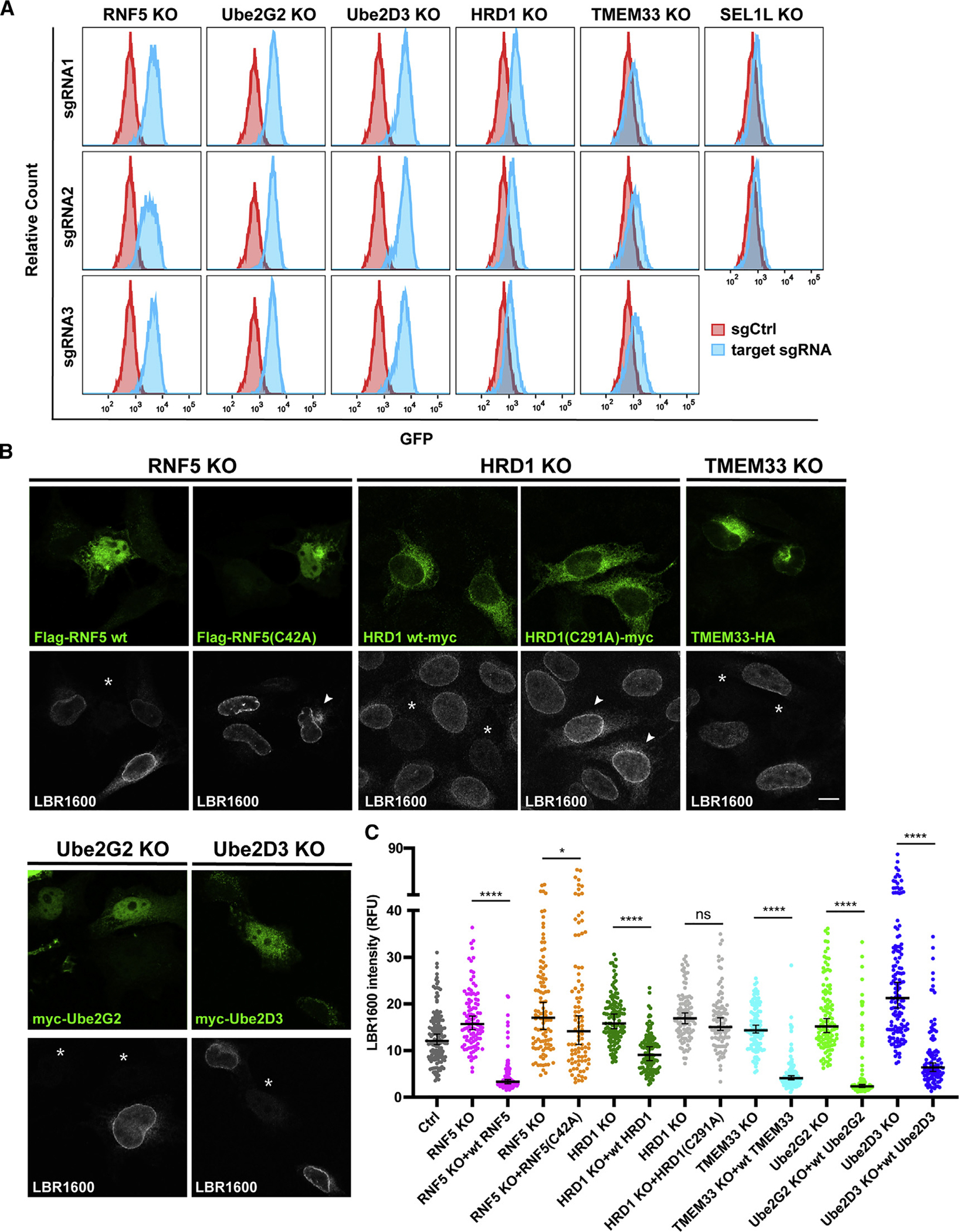
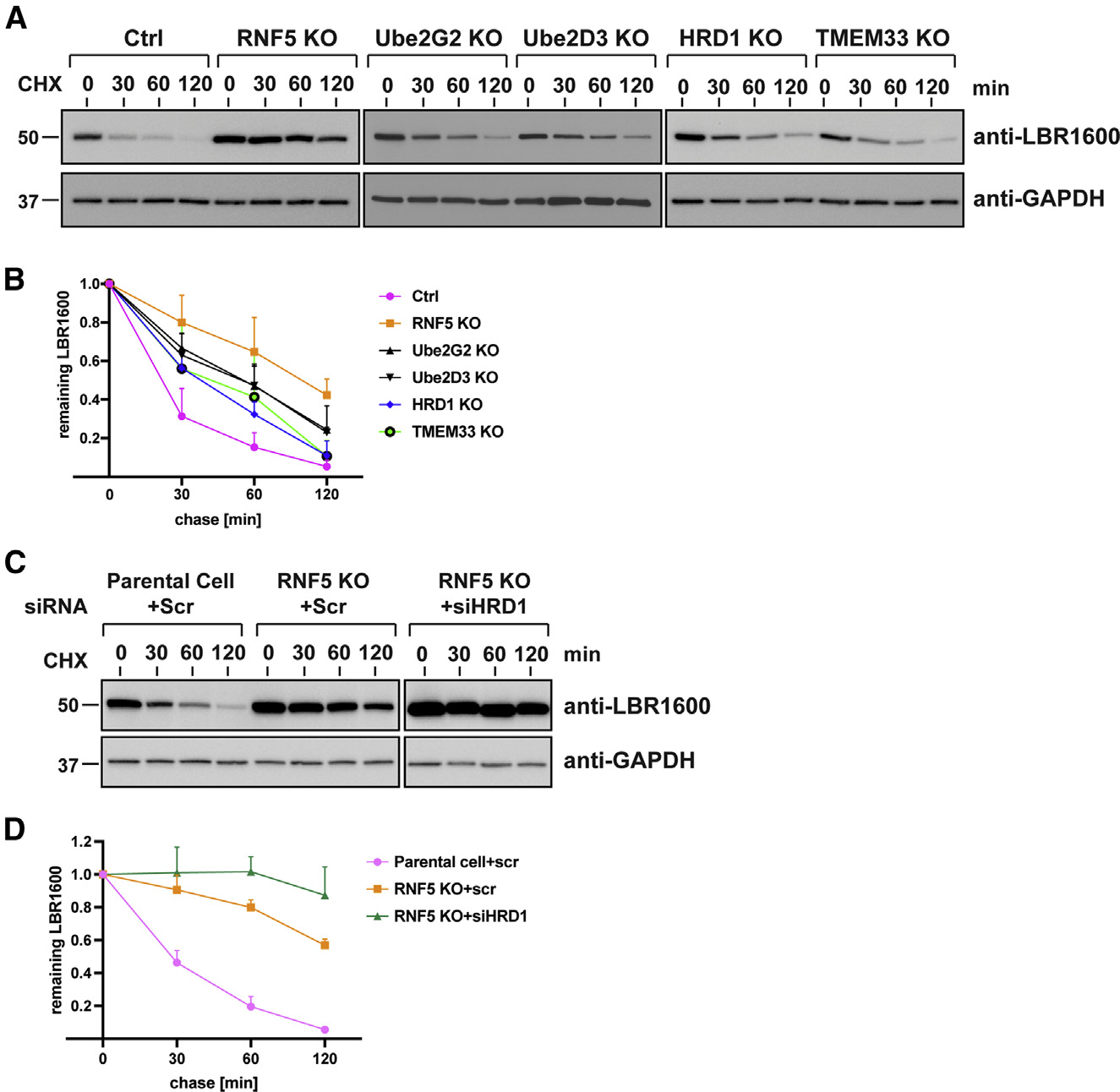
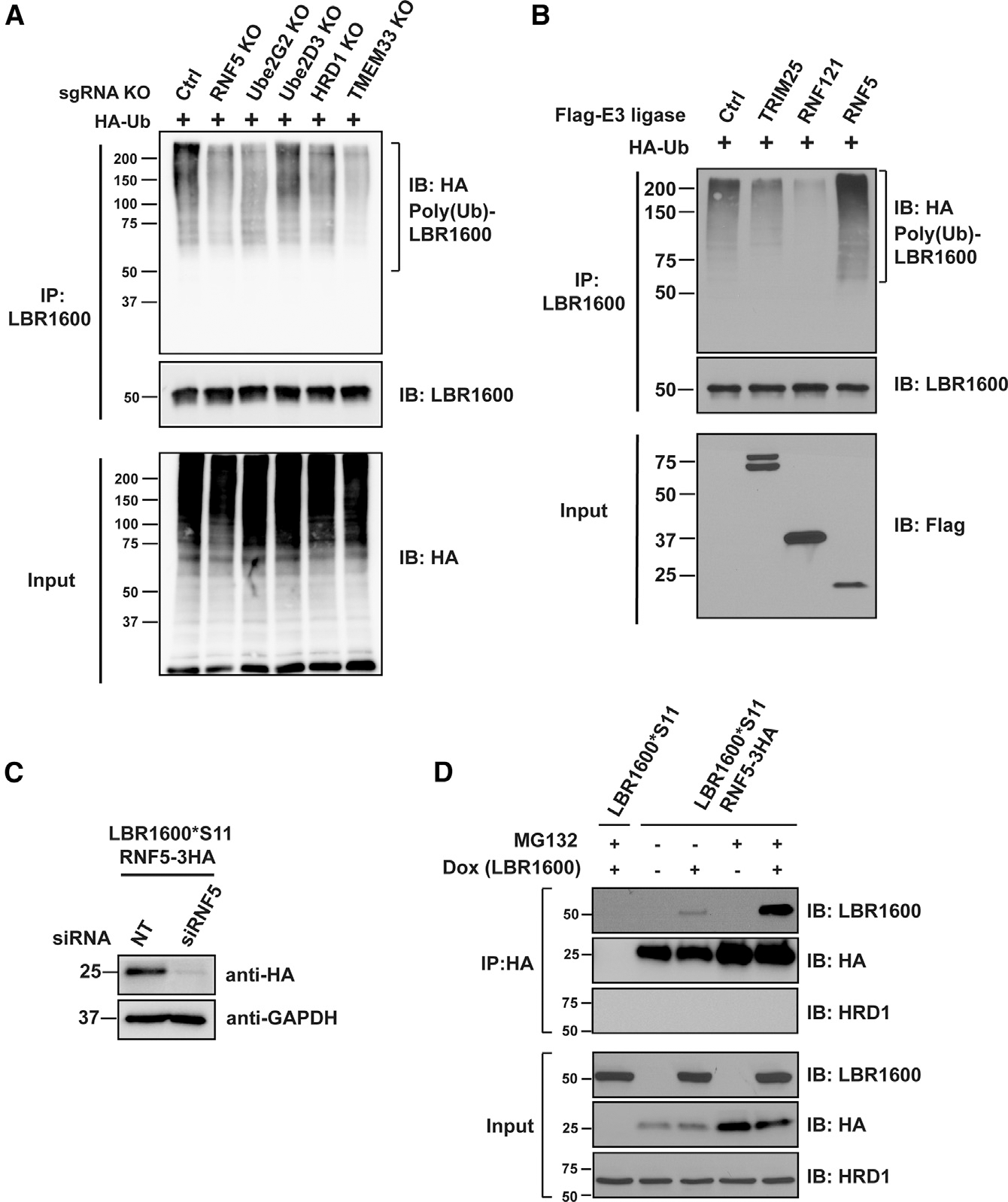
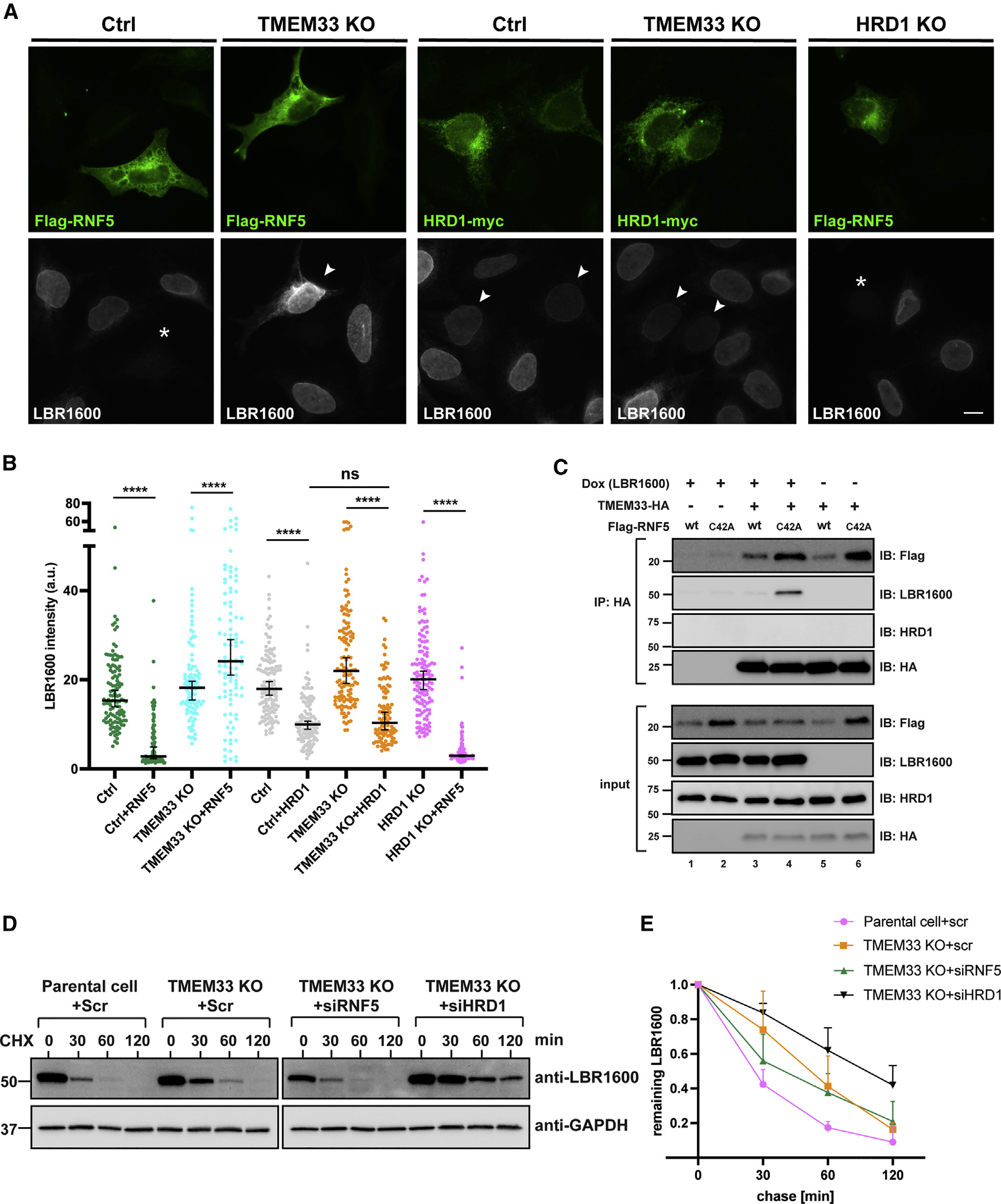
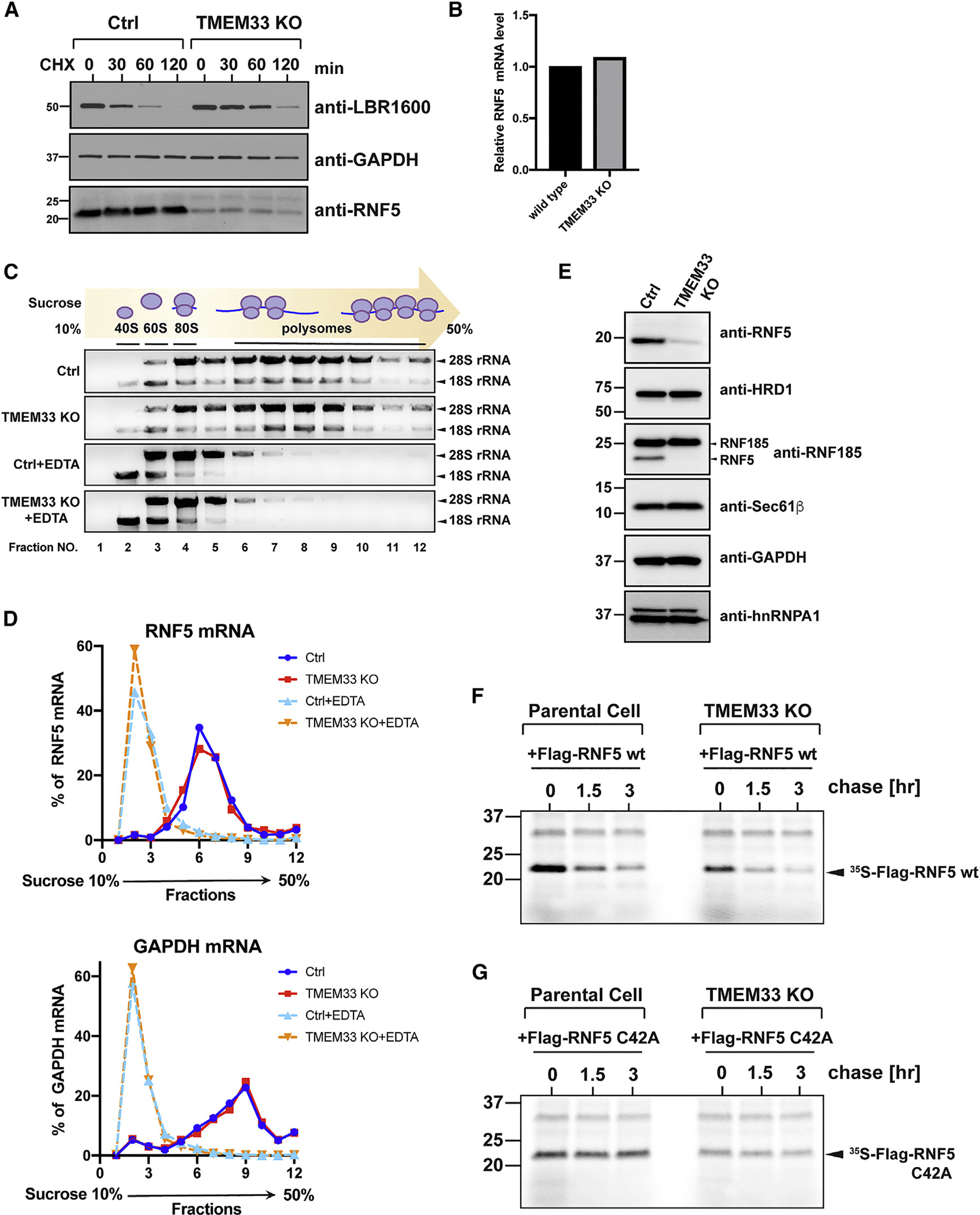
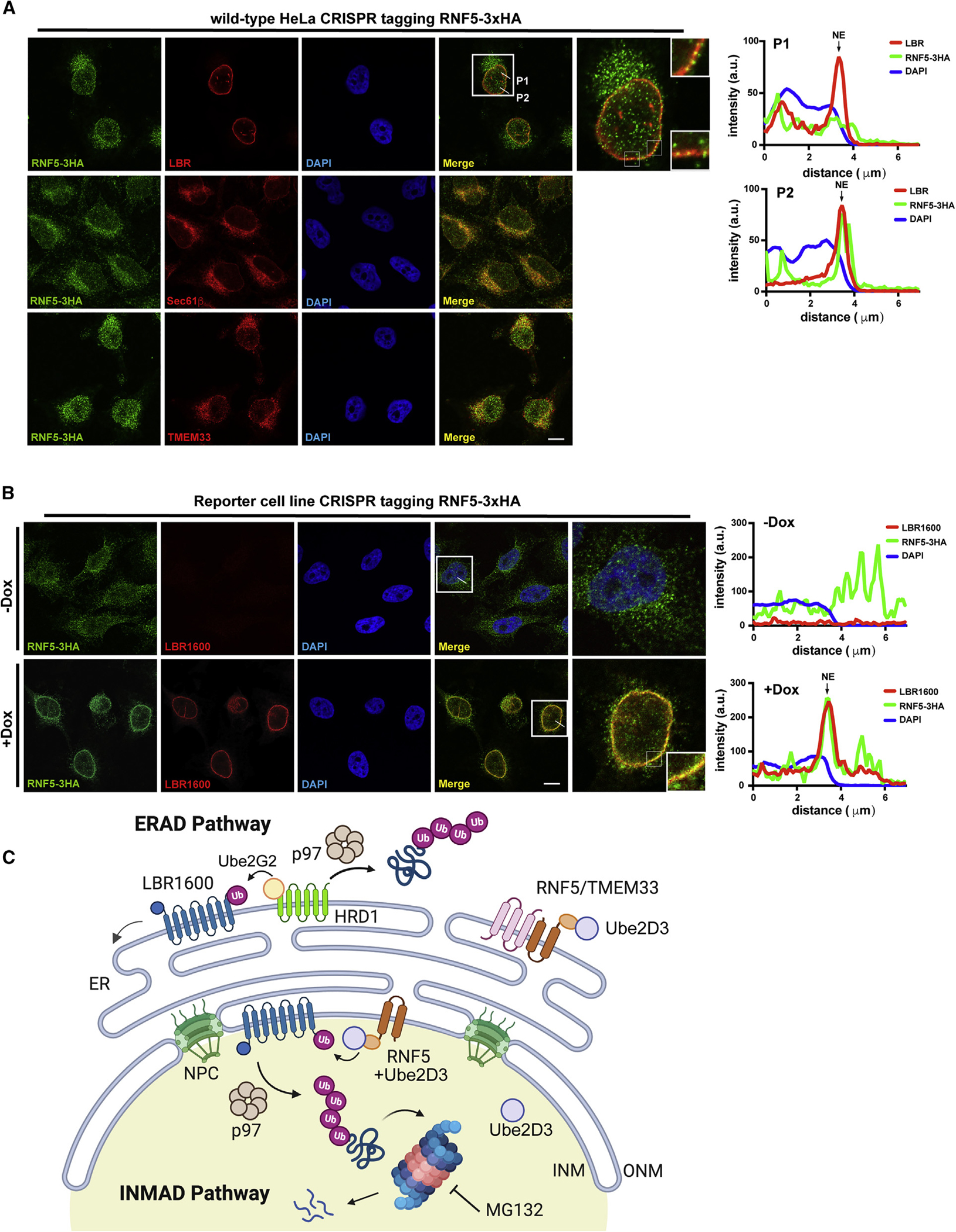
Many human diseases are caused by mutations in nuclear envelope (NE) proteins. How protein homeostasis and disease etiology are interconnected at the NE is poorly understood. Specifically, the identity of local ubiquitin ligases that facilitate ubiquitin-proteasome-dependent NE protein turnover is presently unknown. Here, we employ a short-lived, Lamin B receptor disease variant as a model substrate in a genetic screen to uncover key elements of NE protein turnover. We identify the ubiquitin-conjugating enzymes (E2s) Ube2G2 and Ube2D3, the membrane-resident ubiquitin ligases (E3s) RNF5 and HRD1, and the poorly understood protein TMEM33. RNF5, but not HRD1, requires TMEM33 both for efficient biosynthesis and function. Once synthesized, RNF5 responds dynamically to increased substrate levels at the NE by departing from the endoplasmic reticulum, where HRD1 remains confined. Thus, mammalian protein quality control machinery partitions between distinct cellular compartments to address locally changing substrate loads, establishing a robust cellular quality control system.
Keywords: CP: Molecular biology; CRISPR screen; ERAD; RNF5; TMEM33; nuclear envelopathies; proteasome; protein quality control; protein turnover; proteostasis; ubiquitin ligase.
Copyright © 2022 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
