

RNA-controlled nucleocytoplasmic shuttling of mRNA decay factors regulates mRNA synthesis and a novel mRNA decay pathway
- PMID: 36418294
- PMCID: PMC9684461
- DOI: 10.1038/s41467-022-34417-z
RNA-controlled nucleocytoplasmic shuttling of mRNA decay factors regulates mRNA synthesis and a novel mRNA decay pathway
Abstract
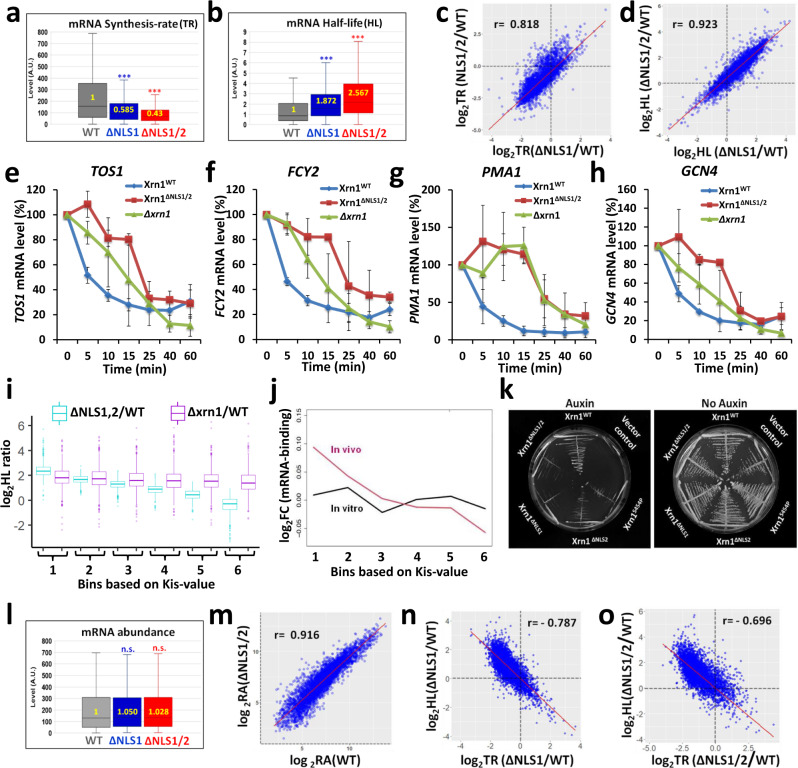
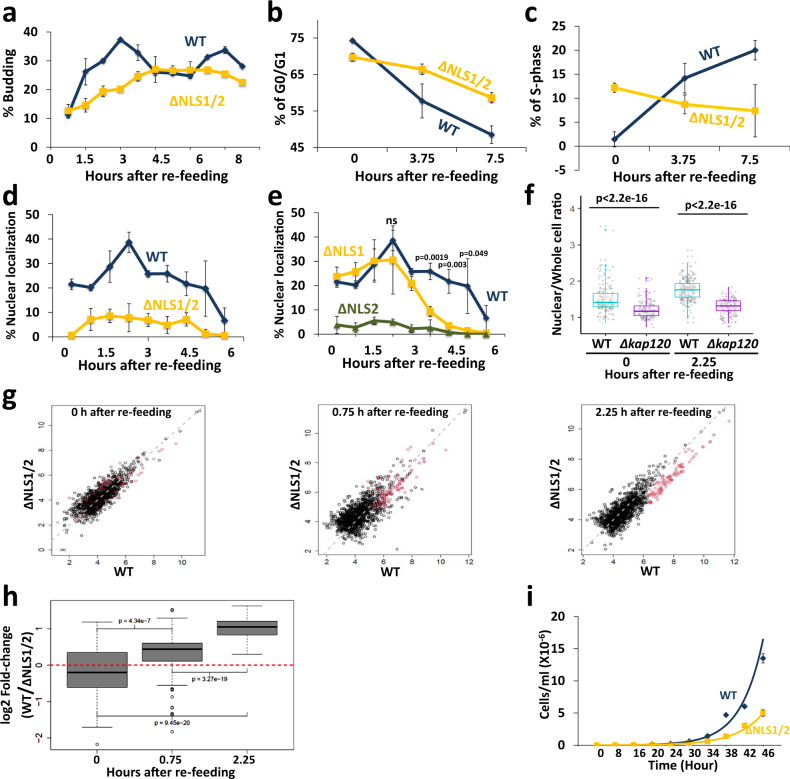
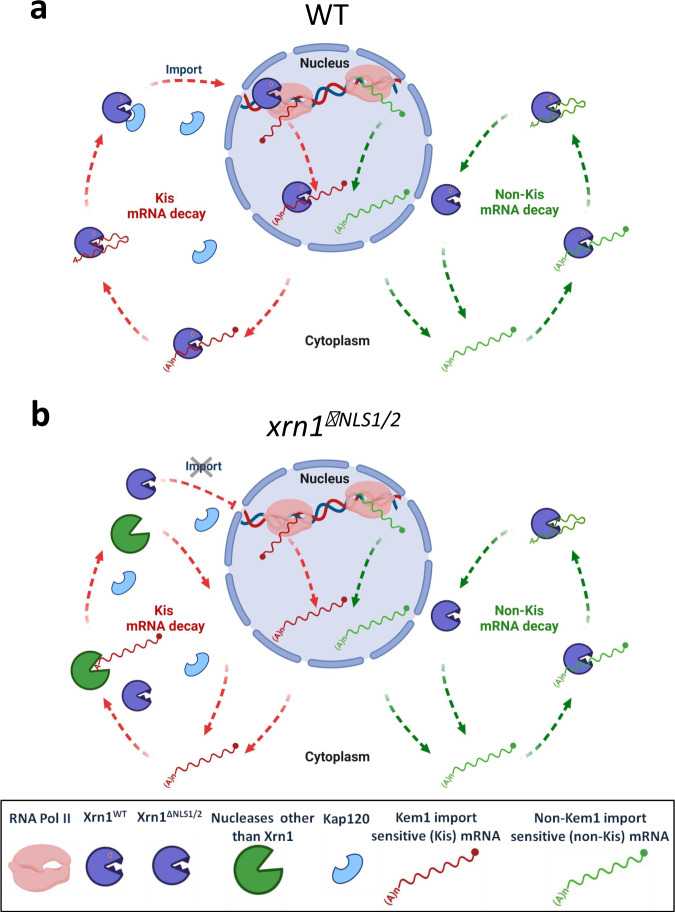
mRNA level is controlled by factors that mediate both mRNA synthesis and decay, including the 5' to 3' exonuclease Xrn1. Here we show that nucleocytoplasmic shuttling of several yeast mRNA decay factors plays a key role in determining both mRNA synthesis and decay. Shuttling is regulated by RNA-controlled binding of the karyopherin Kap120 to two nuclear localization sequences (NLSs) in Xrn1, location of one of which is conserved from yeast to human. The decaying RNA binds and masks NLS1, establishing a link between mRNA decay and Xrn1 shuttling. Preventing Xrn1 import, either by deleting KAP120 or mutating the two Xrn1 NLSs, compromises transcription and, unexpectedly, also cytoplasmic decay, uncovering a cytoplasmic decay pathway that initiates in the nucleus. Most mRNAs are degraded by both pathways - the ratio between them represents a full spectrum. Importantly, Xrn1 shuttling is required for proper responses to environmental changes, e.g., fluctuating temperatures, involving proper changes in mRNA abundance and in cell proliferation rate.
© 2022. The Author(s).
Conflict of interest statement
All authors declare no competing interest.
Figures
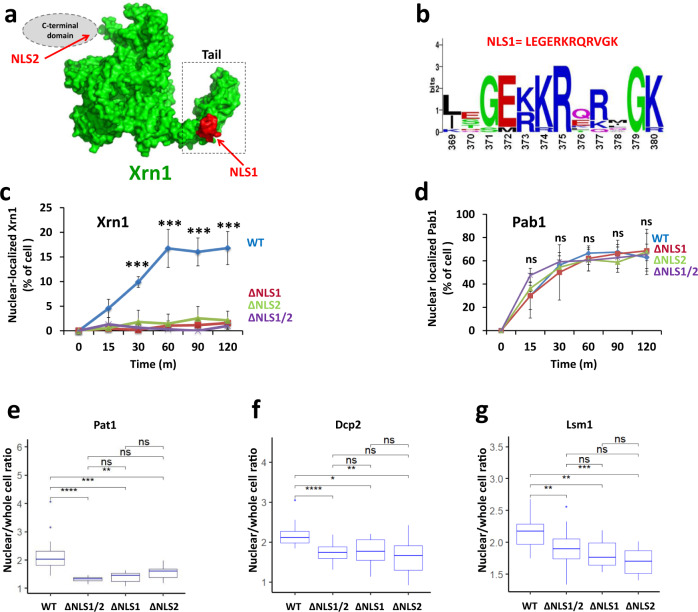
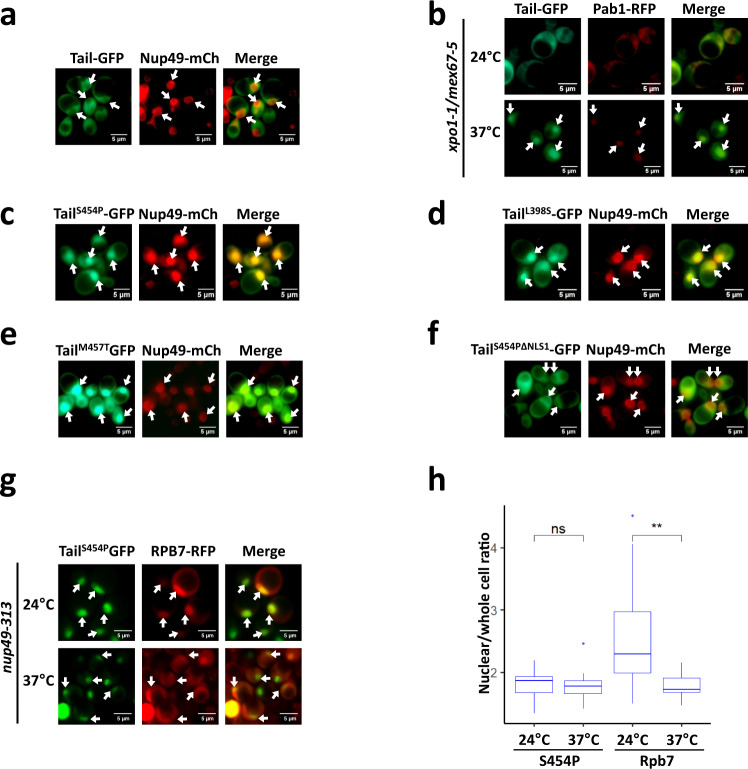
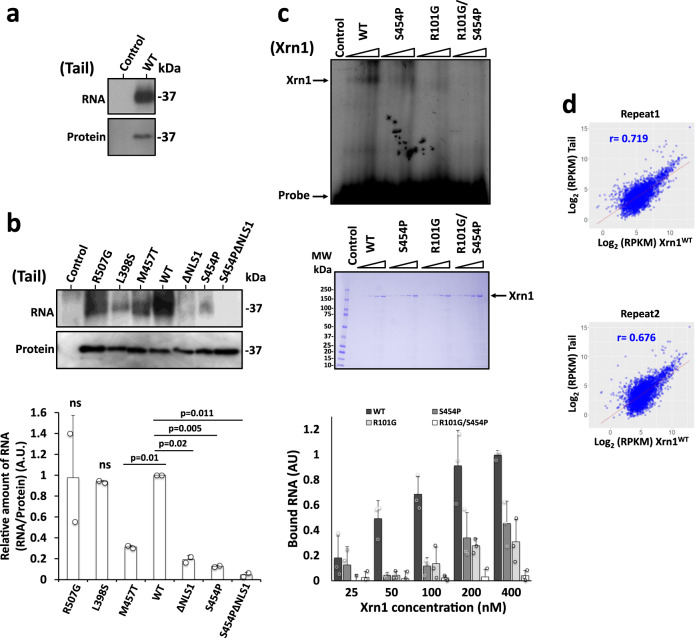
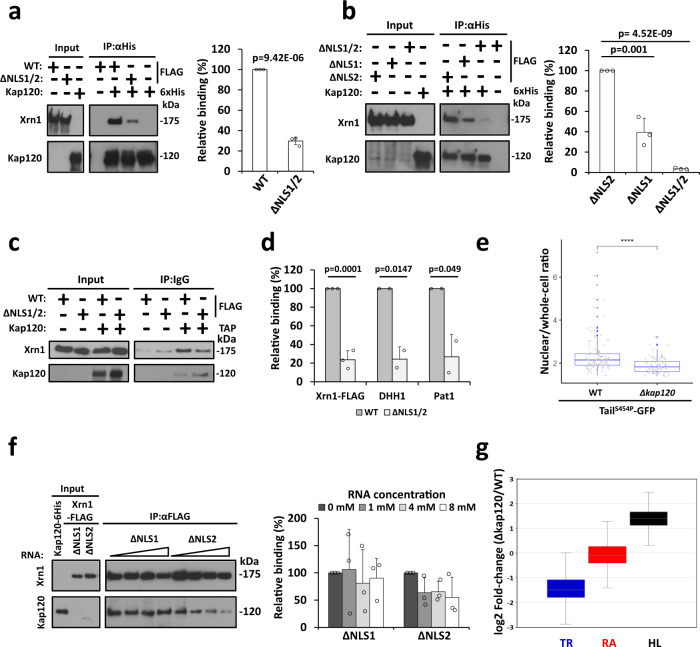
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
