

MEK inhibitor resistance in lung adenocarcinoma is associated with addiction to sustained ERK suppression
- PMID: 36418460
- PMCID: PMC9684561
- DOI: 10.1038/s41698-022-00328-x
MEK inhibitor resistance in lung adenocarcinoma is associated with addiction to sustained ERK suppression
Abstract
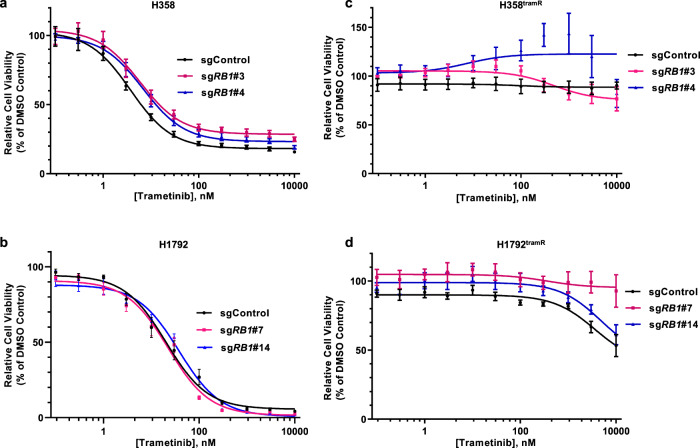
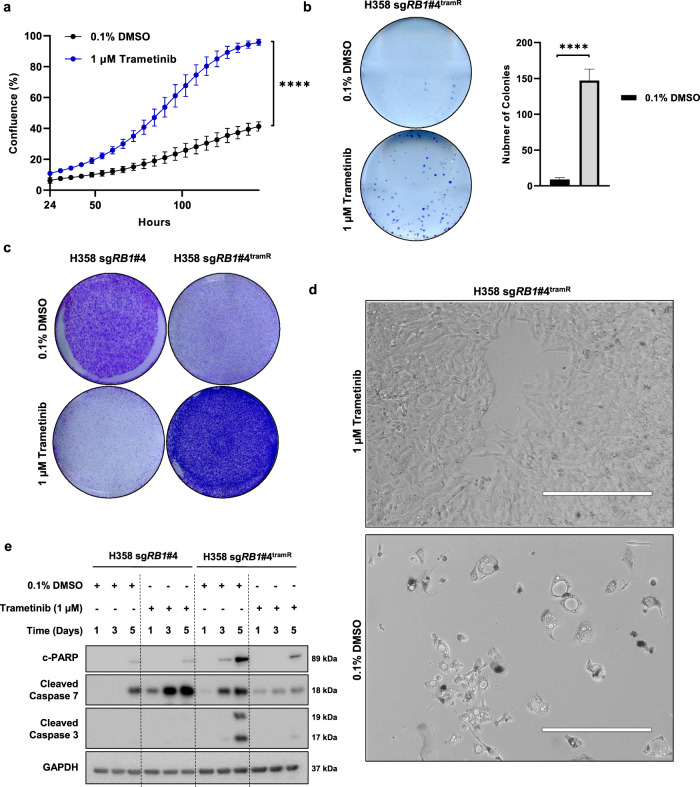
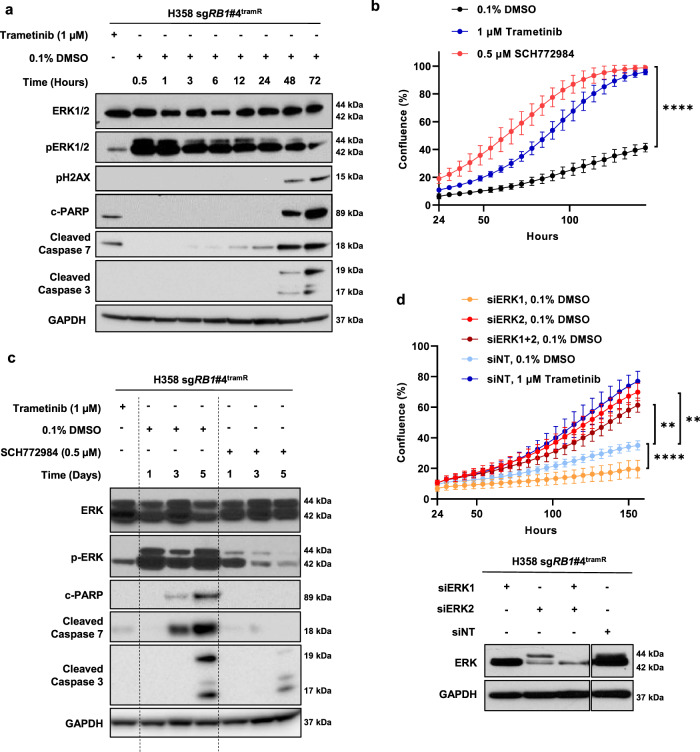
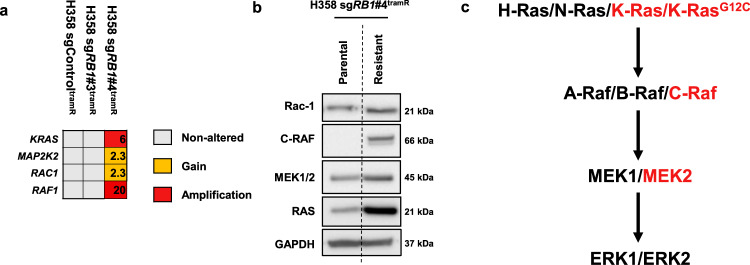
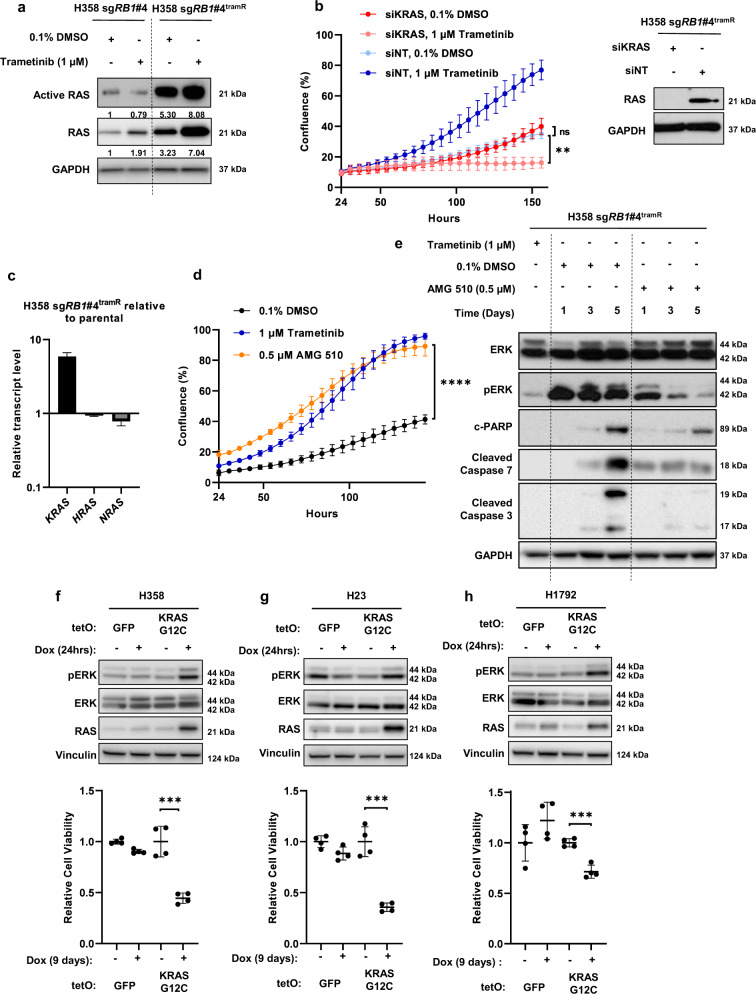
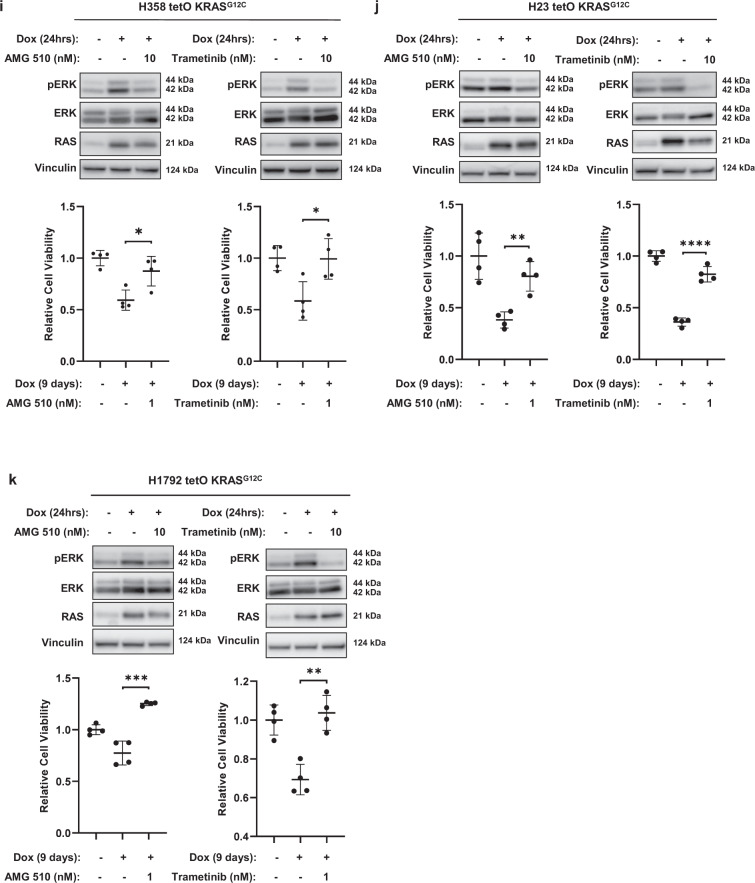
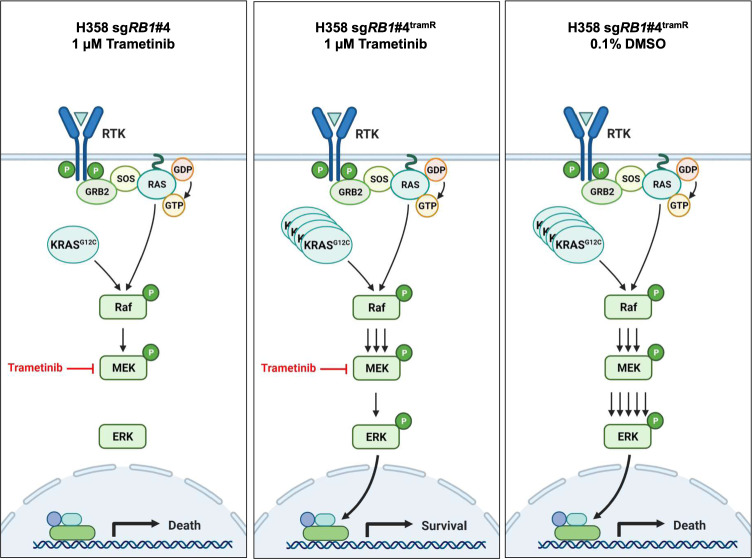
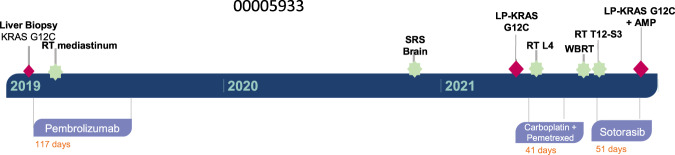
MEK inhibitors (MEKi) have limited efficacy in KRAS mutant lung adenocarcinoma (LUAD) patients, and this is attributed to both intrinsic and adaptive mechanisms of drug resistance. While many studies have focused on the former, there remains a dearth of data regarding acquired resistance to MEKi in LUAD. We established trametinib-resistant KRAS mutant LUAD cells through dose escalation and performed targeted MSK-IMPACT sequencing to identify drivers of MEKi resistance. Comparing resistant cells to their sensitive counterparts revealed alteration of genes associated with trametinib response. We describe a state of "drug addiction" in resistant cases where cells are dependent on continuous culture in trametinib for survival. We show that dependence on ERK2 suppression underlies this phenomenon and that trametinib removal hyperactivates ERK, resulting in ER stress and apoptosis. Amplification of KRASG12C occurs in drug-addicted cells and blocking mutant-specific activity with AMG 510 rescues the lethality associated with trametinib withdrawal. Furthermore, we show that increased KRASG12C expression is lethal to other KRAS mutant LUAD cells, consequential to ERK hyperactivation. Our study determines the drug-addicted phenotype in lung cancer is associated with KRAS amplification and demonstrates that toxic acquired genetic changes can develop de novo in the background of MAPK suppression with MEK inhibitors. We suggest that the presence of mutant KRAS amplification in patients may identify those that may benefit from a "drug holiday" to circumvent drug resistance. These findings demonstrate the toxic potential of hyperactive ERK signaling and highlight potential therapeutic opportunities in patients bearing KRAS mutations.
© 2022. The Author(s).
Conflict of interest statement
A.M.U. and W.W.L. are scientific advisors for Hyperbio Therapeutics. Marc Ladanyi has received advisory board compensation from Merck, Bristol-Myers Squibb, Takeda, Bayer, Lilly Oncology, Janssen, and Paige.AI. Research grants unrelated to the current study was obtained from Hesinn Heathcare, Merus, LOXO Oncology and Elevation Oncology Inc. Romel Somwar has received research grants, unrelated to the current study, from Merus, LOXO Oncology and Elevation Oncology Inc. and Helsinn.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
