

The genetic and biochemical determinants of mRNA degradation rates in mammals
- PMID: 36419176
- PMCID: PMC9684954
- DOI: 10.1186/s13059-022-02811-x
The genetic and biochemical determinants of mRNA degradation rates in mammals
Abstract
Background: Degradation rate is a fundamental aspect of mRNA metabolism, and the factors governing it remain poorly characterized. Understanding the genetic and biochemical determinants of mRNA half-life would enable more precise identification of variants that perturb gene expression through post-transcriptional gene regulatory mechanisms.
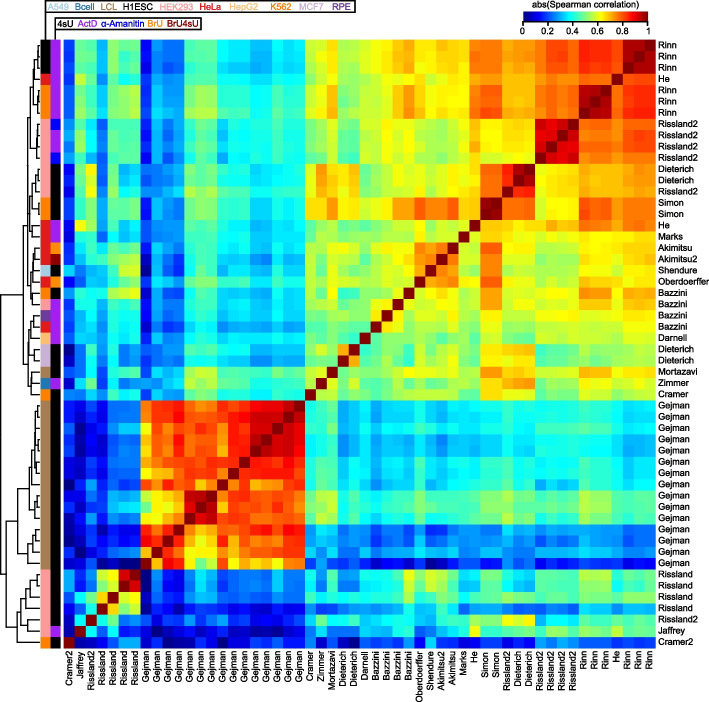
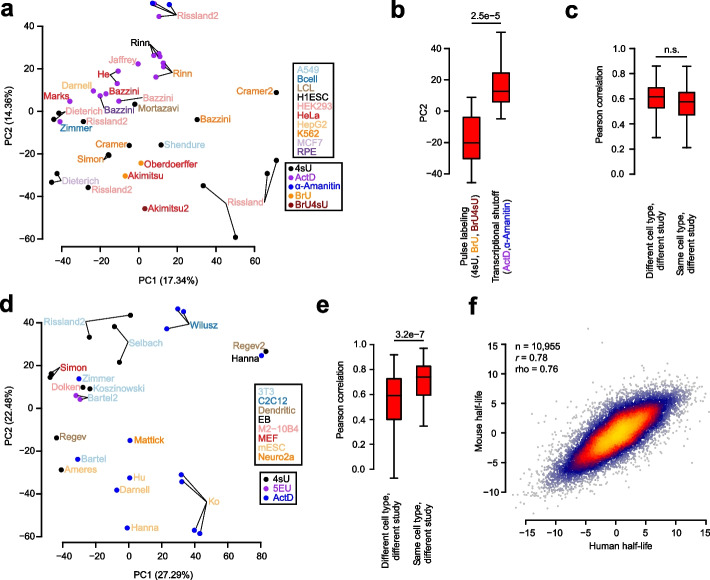
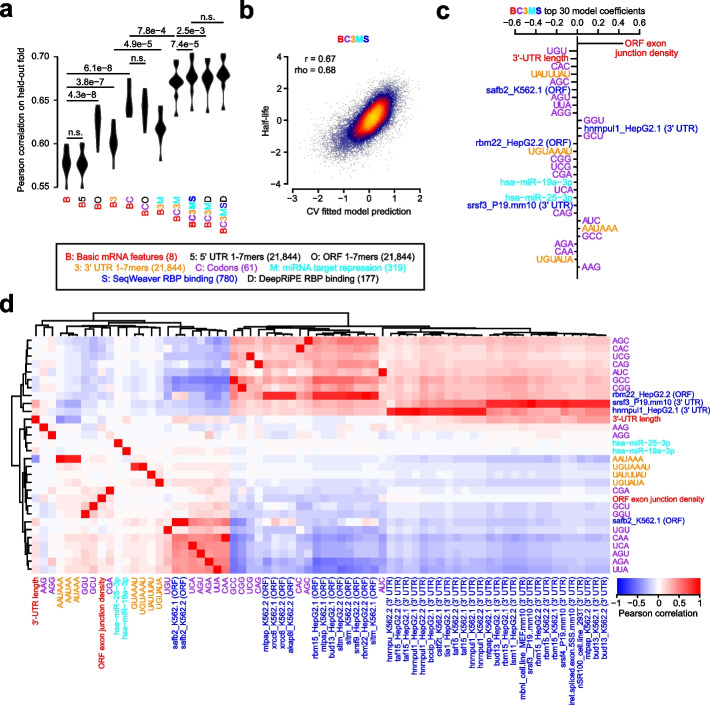
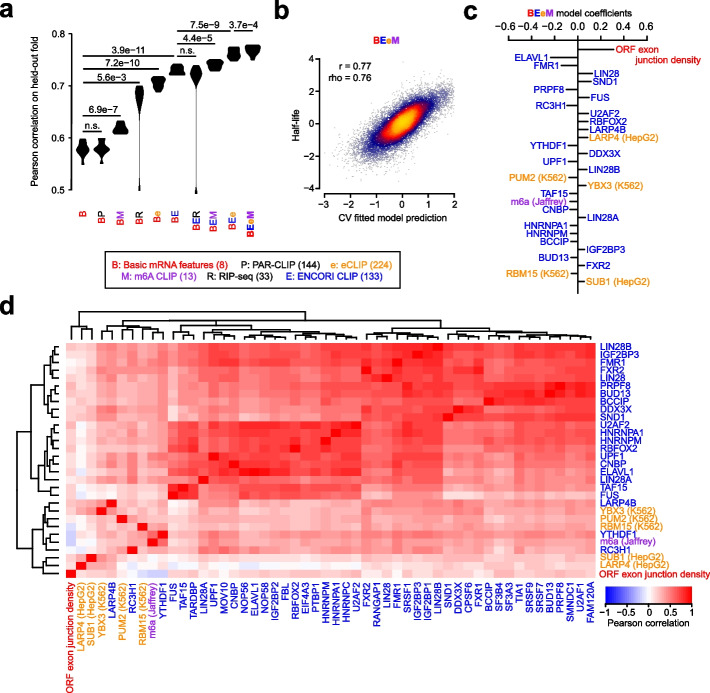
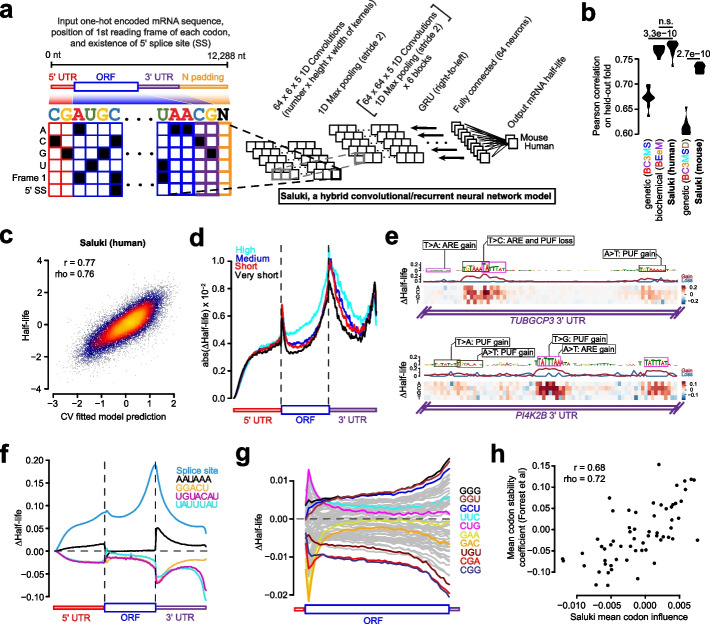
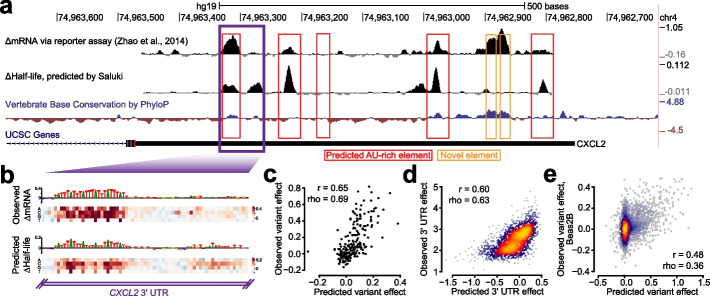
Results: We establish a compendium of 39 human and 27 mouse transcriptome-wide mRNA decay rate datasets. A meta-analysis of these data identified a prevalence of technical noise and measurement bias, induced partially by the underlying experimental strategy. Correcting for these biases allowed us to derive more precise, consensus measurements of half-life which exhibit enhanced consistency between species. We trained substantially improved statistical models based upon genetic and biochemical features to better predict half-life and characterize the factors molding it. Our state-of-the-art model, Saluki, is a hybrid convolutional and recurrent deep neural network which relies only upon an mRNA sequence annotated with coding frame and splice sites to predict half-life (r=0.77). The key novel principle learned by Saluki is that the spatial positioning of splice sites, codons, and RNA-binding motifs within an mRNA is strongly associated with mRNA half-life. Saluki predicts the impact of RNA sequences and genetic mutations therein on mRNA stability, in agreement with functional measurements derived from massively parallel reporter assays.
Conclusions: Our work produces a more robust ground truth for transcriptome-wide mRNA half-lives in mammalian cells. Using these revised measurements, we trained Saluki, a model that is over 50% more accurate in predicting half-life from sequence than existing models. Saluki succinctly captures many of the known determinants of mRNA half-life and can be rapidly deployed to predict the functional consequences of arbitrary mutations in the transcriptome.
Keywords: Deep neural networks; Post-transcriptional gene regulation; mRNA half-life; mRNA stability.
© 2022. The Author(s).
Conflict of interest statement
V.A. and D.R.K. are employees of Calico Life Sciences.
Figures
References
-
- Agarwal V, Shendure J. Predicting mRNA abundance directly from genomic sequence using deep convolutional neural networks. Cell Rep. 2020;31:107663. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
