

Characterization of the Complete Mitochondrial Genome of the Spotted Catfish Arius maculatus (Thunberg, 1792) and Its Phylogenetic Implications
- PMID: 36421803
- PMCID: PMC9690425
- DOI: 10.3390/genes13112128
Characterization of the Complete Mitochondrial Genome of the Spotted Catfish Arius maculatus (Thunberg, 1792) and Its Phylogenetic Implications
Abstract
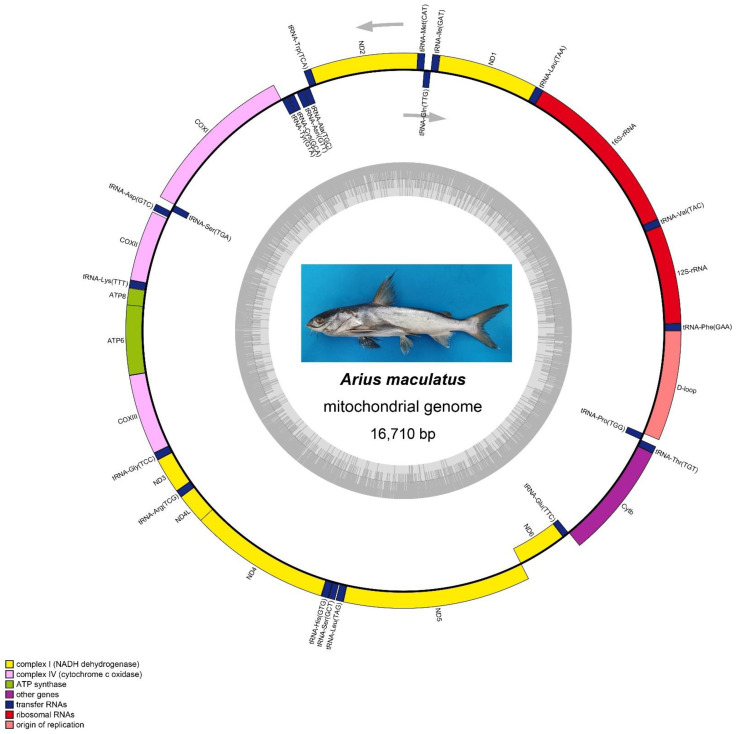
The spotted catfish, Arius maculatus (Siluriformes), is an important economical aquaculture species inhabiting the Indian Ocean, as well as the western Pacific Ocean. The bioinformatics data in previous studies about the phylogenetic reconstruction of Siluriformes were insufficient and incomplete. In the present study, we presented a newly sequenced A. maculatus mitochondrial genome (mtDNA). The A. maculatus mtDNA was 16,710 bp in length and contained two ribosomal RNA (rRNA) genes, thirteen protein-coding genes (PCGs), twenty-two transfer RNA (tRNA) genes, and one D-loop region. The composition and order of these above genes were similar to those found in most other vertebrates. The relative synonymous codon usage (RSCU) of the 13 PCGs in A. maculatus mtDNA was consistent with that of PCGs in other published Siluriformes mtDNA. Furthermore, the average non-synonymous/synonymous mutation ratio (Ka/Ks) analysis, based on the 13 PCGs of the four Ariidae species, showed a strong purifying selection. Additionally, phylogenetic analysis, according to 13 concatenated PCG nucleotide and amino acid datasets, showed that A. maculatus and Netuma thalassina (Netuma), Occidentarius platypogon (Occidentarius), and Bagre panamensis (Bagre) were clustered as sister clade. The complete mtDNA of A. maculatus provides a helpful dataset for research on the population structure and genetic diversity of Ariidae species.
Keywords: Arius maculatus; Ka/Ks; RSCU; mitochondrial genome; phylogenetic analysis.
Conflict of interest statement
The authors report no conflict of interest.
Figures






Similar articles
-
Molecular characterization and phylogenetic analyses of the mitogenome of Wan-Xi white goose, a native goose breed in China.BMC Genom Data. 2025 May 13;26(1):34. doi: 10.1186/s12863-025-01326-1. BMC Genom Data. 2025. PMID: 40360978 Free PMC article.
-
Characterization of complete mitochondrial genome of fives tripe wrasse (Thalassoma quinquevittatum, Lay & Bennett, 1839) and phylogenetic analysis.Gene. 2017 Jan 20;598:71-78. doi: 10.1016/j.gene.2016.10.042. Epub 2016 Nov 2. Gene. 2017. PMID: 27816474
-
Sequencing and characterization of the complete mitochondrial genome of Japanese Swellshark (Cephalloscyllium umbratile).Sci Rep. 2017 Nov 10;7(1):15299. doi: 10.1038/s41598-017-15702-0. Sci Rep. 2017. PMID: 29127415 Free PMC article.
-
Characterization of the complete mitochondrial genome of Macrotocinclus affinis (Siluriformes; Loricariidae) and phylogenetic studies of Siluriformes.Mol Biol Rep. 2021 Jan;48(1):677-689. doi: 10.1007/s11033-020-06120-z. Epub 2021 Jan 13. Mol Biol Rep. 2021. PMID: 33442829
-
Complete mitochondrial genome of the bullhead torrent catfish, Liobagrus obesus (Siluriformes, Amblycipididae): Genome description and phylogenetic considerations inferred from the Cyt b and 16S rRNA genes.Gene. 2007 Jul 1;396(1):13-27. doi: 10.1016/j.gene.2007.01.027. Epub 2007 Feb 12. Gene. 2007. PMID: 17434693
Cited by
-
The Complete Mitochondrial Genome of Liobagrus huaiheensis (Teleostei: Siluriformes: Amblycipitidae): Characterization, Phylogenetic Placement, and Insights into Genetic Diversity.Genes (Basel). 2025 Aug 19;16(8):977. doi: 10.3390/genes16080977. Genes (Basel). 2025. PMID: 40870025 Free PMC article.
-
Mitochondrial genome of eight Carangidae and phylogenetic analysis in the family.PLoS One. 2025 Jul 9;20(7):e0326619. doi: 10.1371/journal.pone.0326619. eCollection 2025. PLoS One. 2025. PMID: 40632726 Free PMC article.
-
Complete Mitochondrial Genomes of Nannostomus Pencilfish: Genome Characterization and Phylogenetic Analysis.Animals (Basel). 2024 May 29;14(11):1598. doi: 10.3390/ani14111598. Animals (Basel). 2024. PMID: 38891645 Free PMC article.
References
-
- Lei R., Shore G.D., Brenneman R.A., Engberg S.E., Sitzmann B.D., Bailey C.A., Kimmel L.M., Randriamampionona R., Ranaivoarisoa J.F., Louis E.J. Complete sequence and gene organization of the mitochondrial genome for Hubbard′s sportive lemur (Lepilemur hubbardorum) Gene. 2010;464:44–49. doi: 10.1016/j.gene.2010.06.001. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
