

Pathophysiology and pathogenic mechanisms of pulmonary hypertension: role of membrane receptors, ion channels, and Ca2+ signaling
- PMID: 36422993
- PMCID: PMC10110735
- DOI: 10.1152/physrev.00030.2021
Pathophysiology and pathogenic mechanisms of pulmonary hypertension: role of membrane receptors, ion channels, and Ca2+ signaling
Abstract
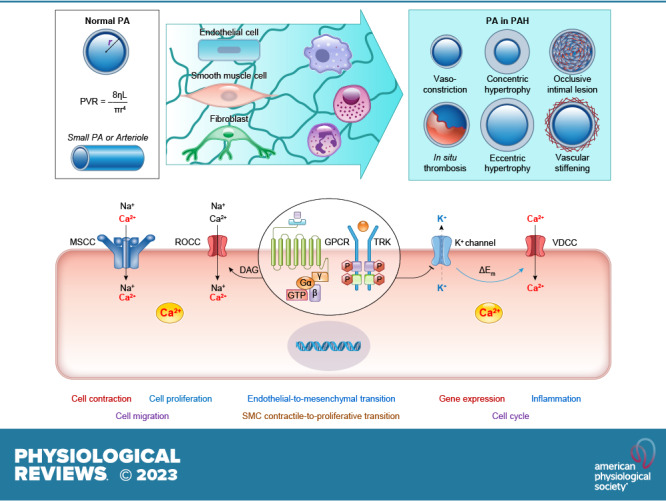
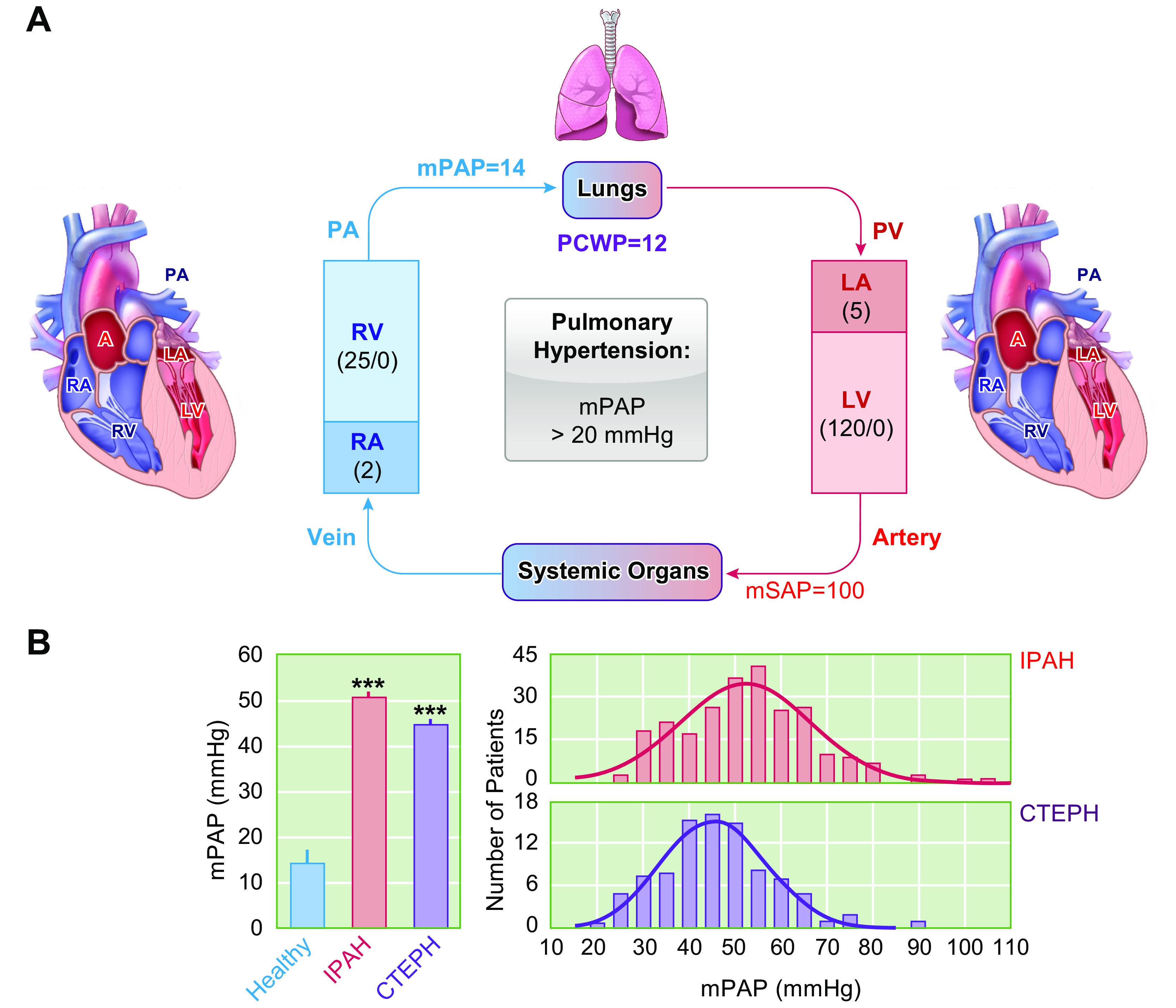
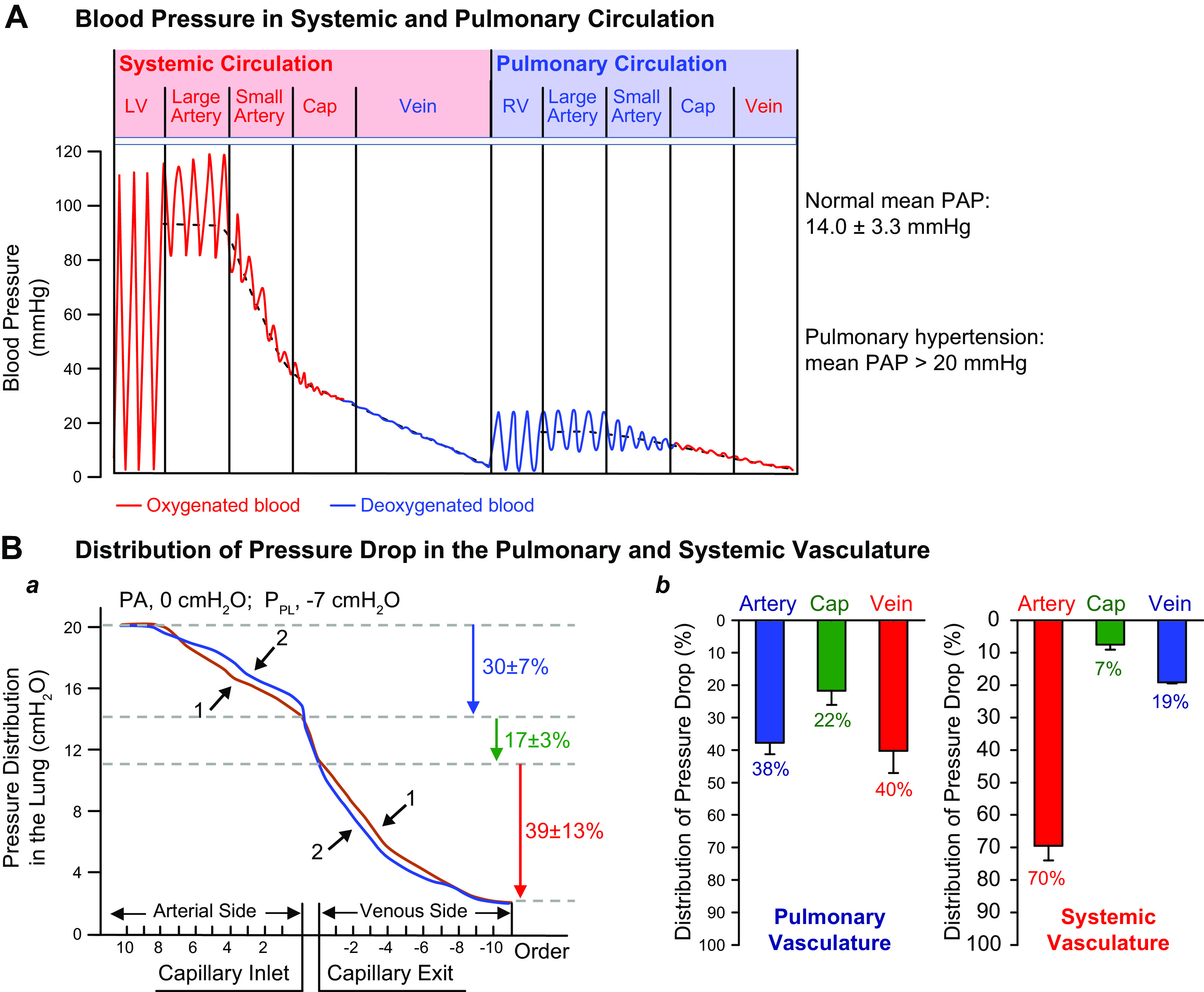
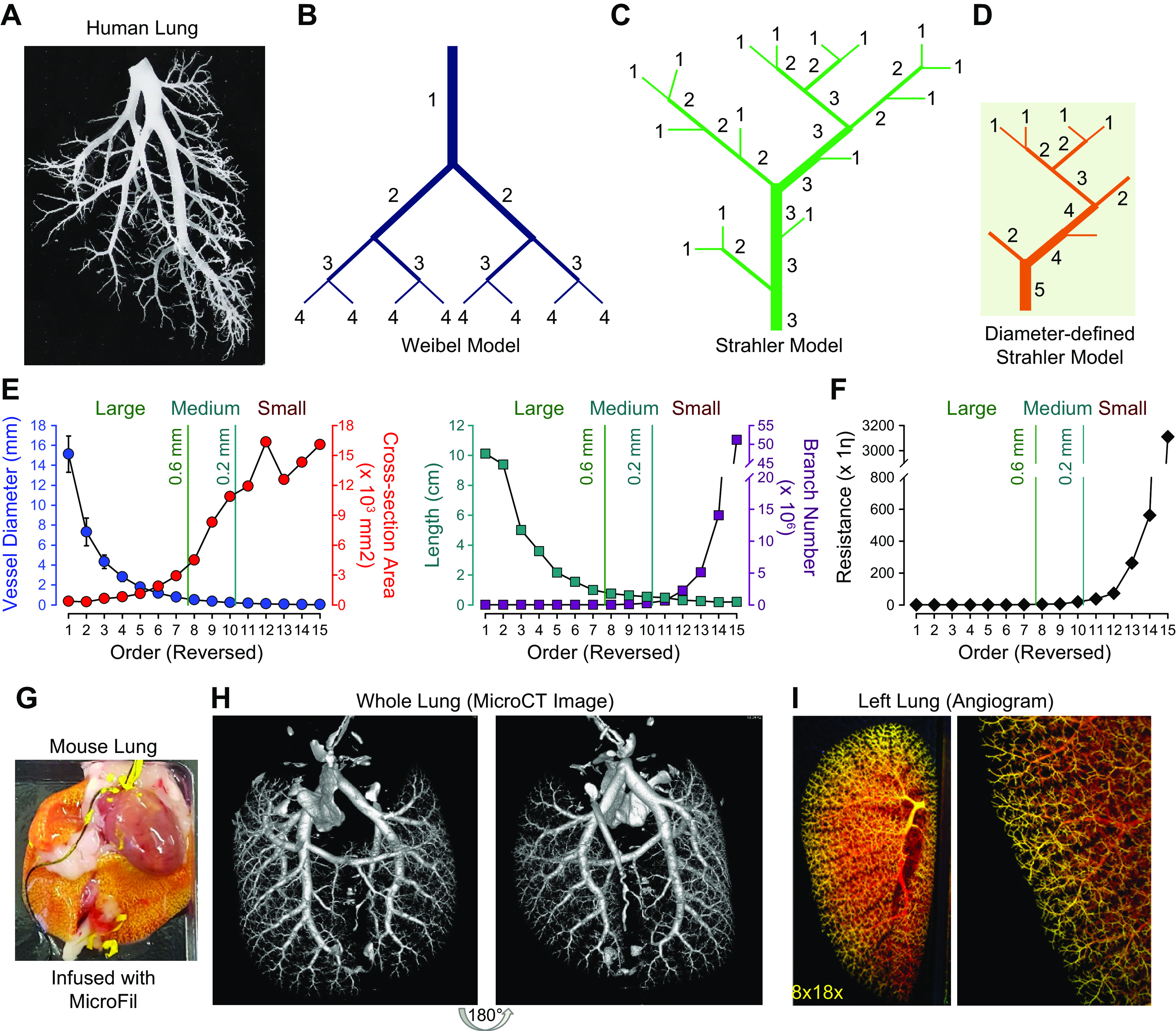
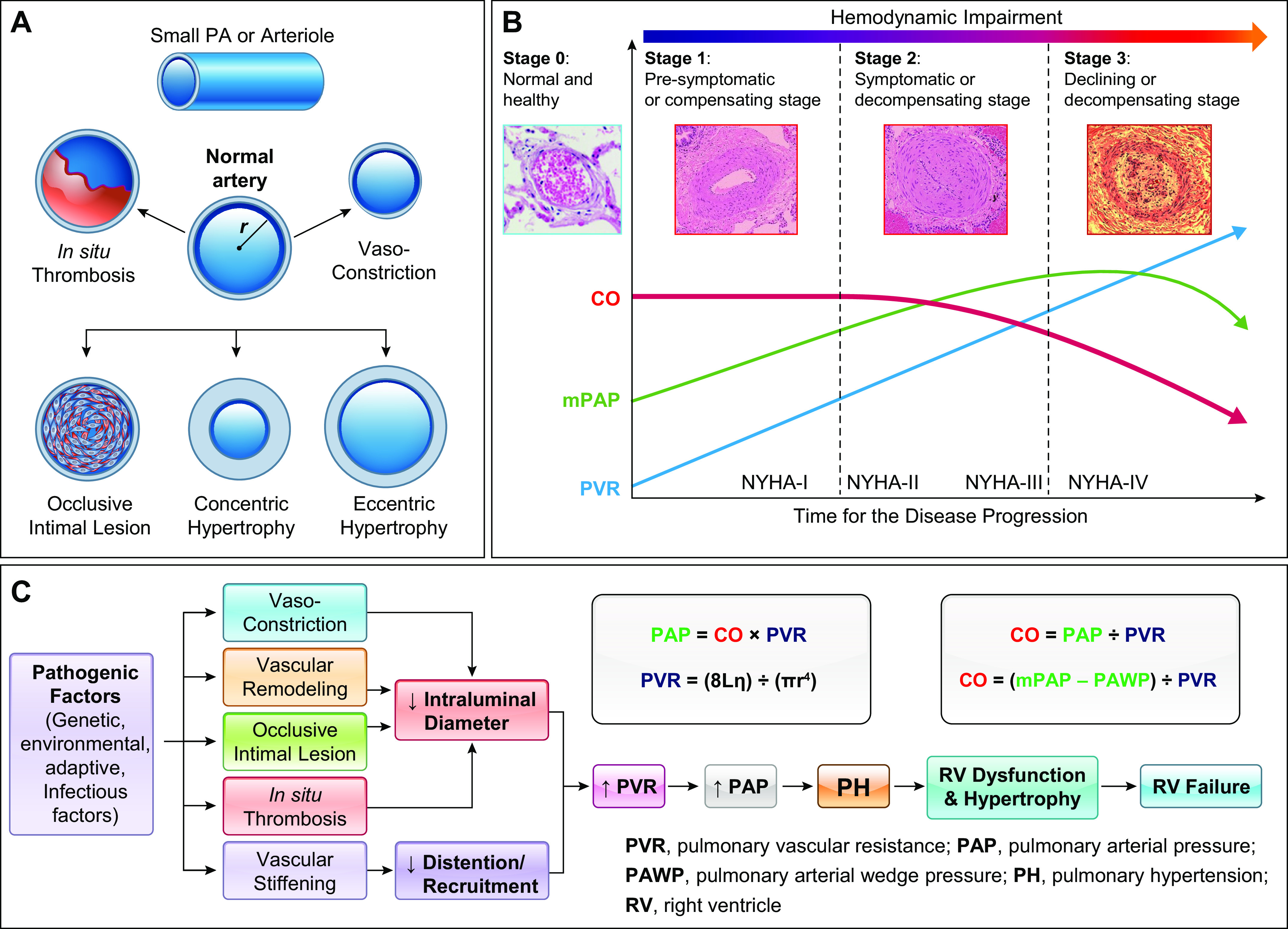
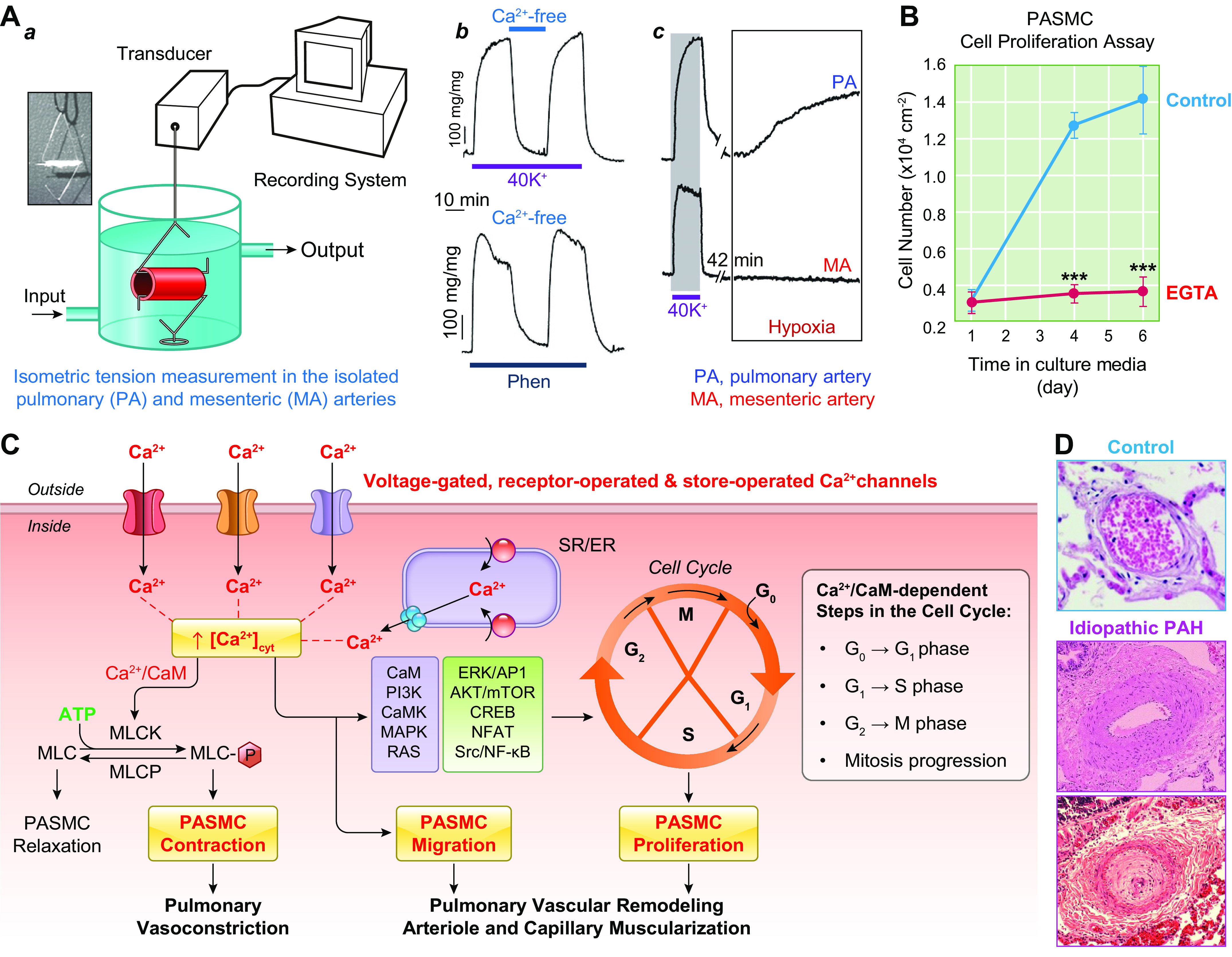
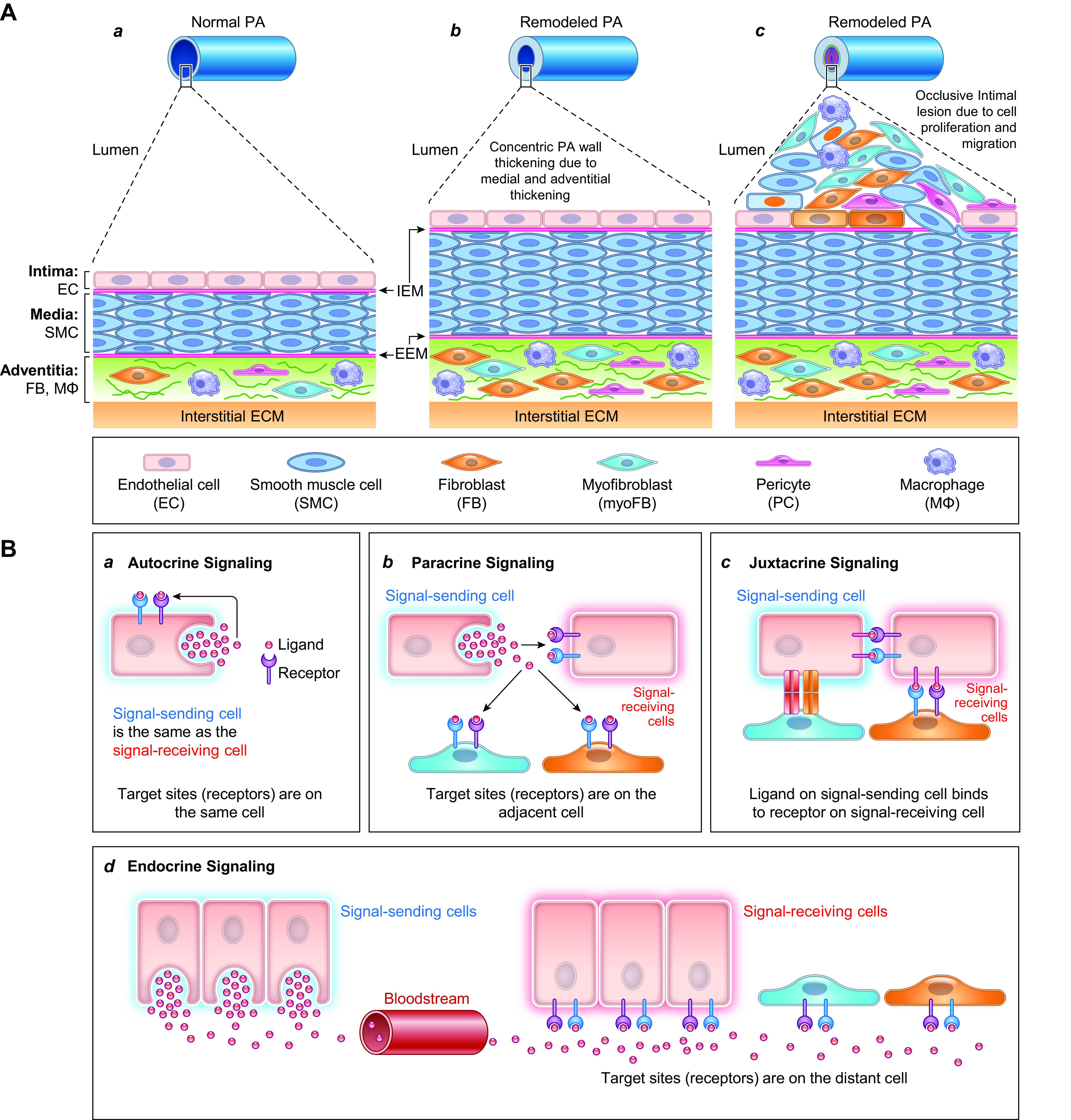
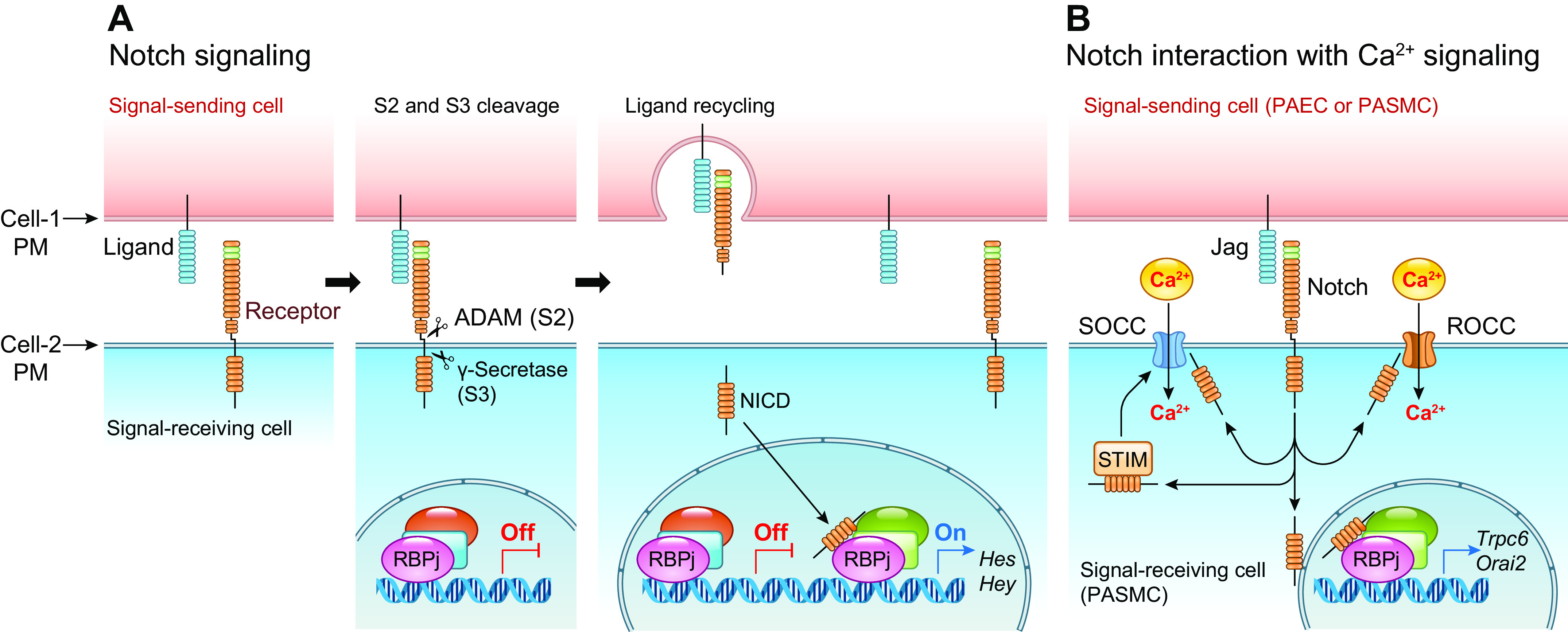
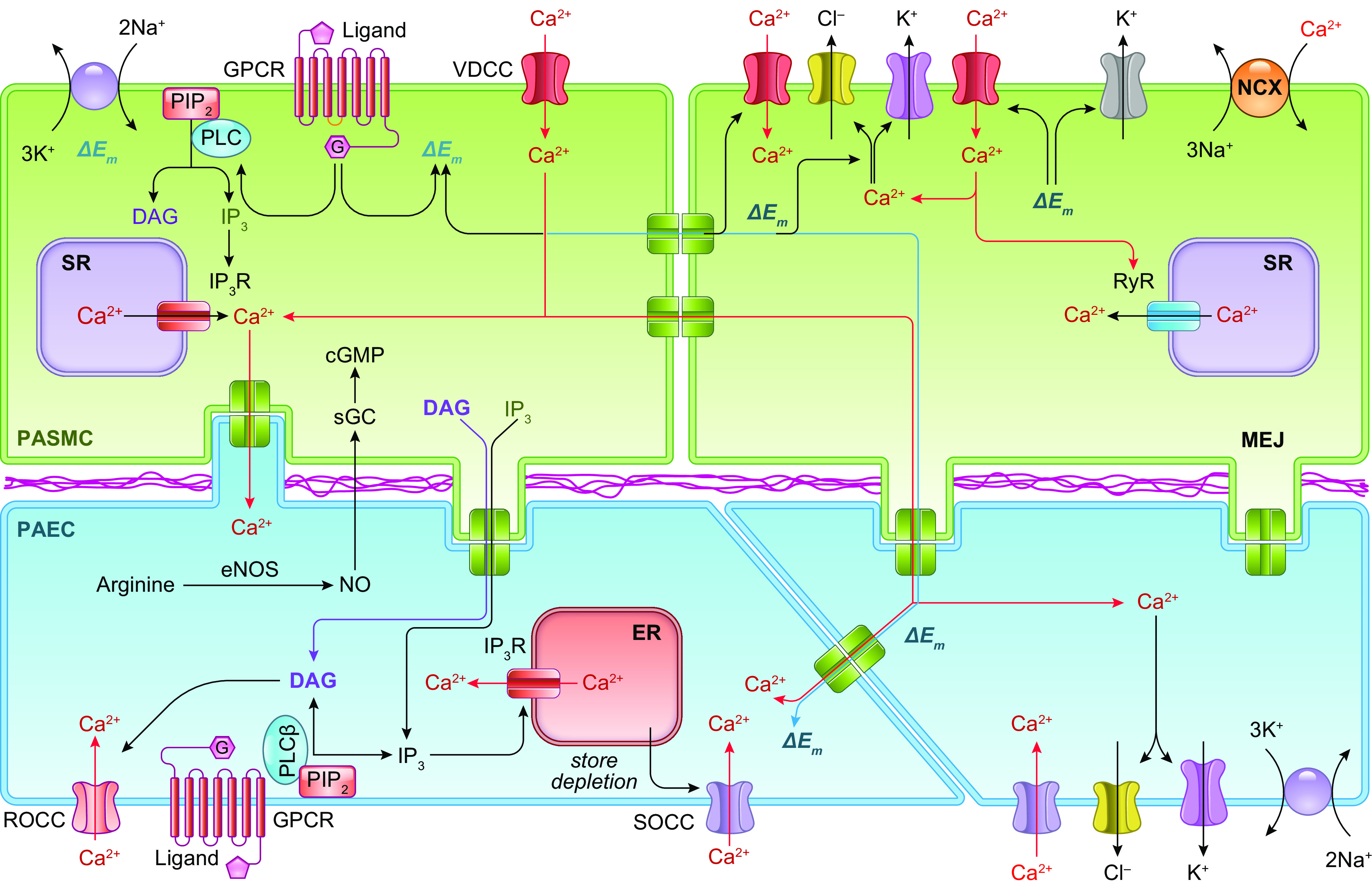
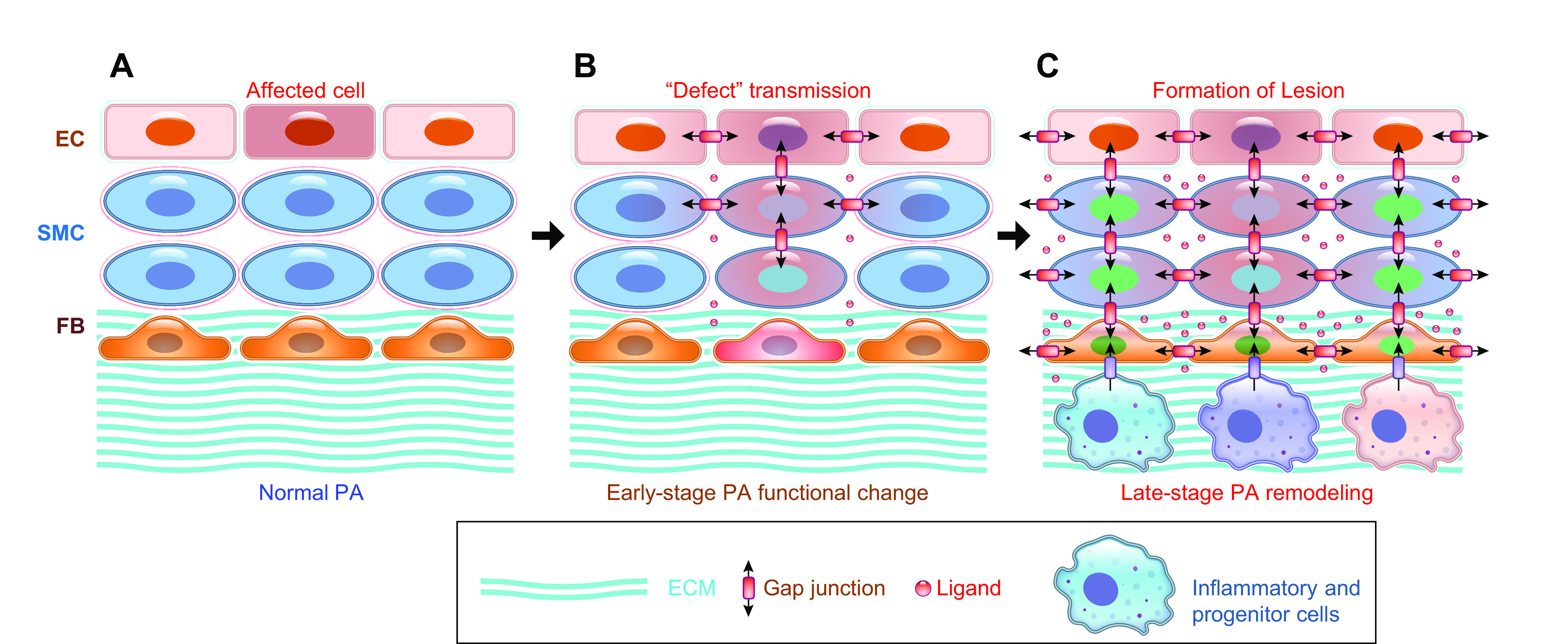
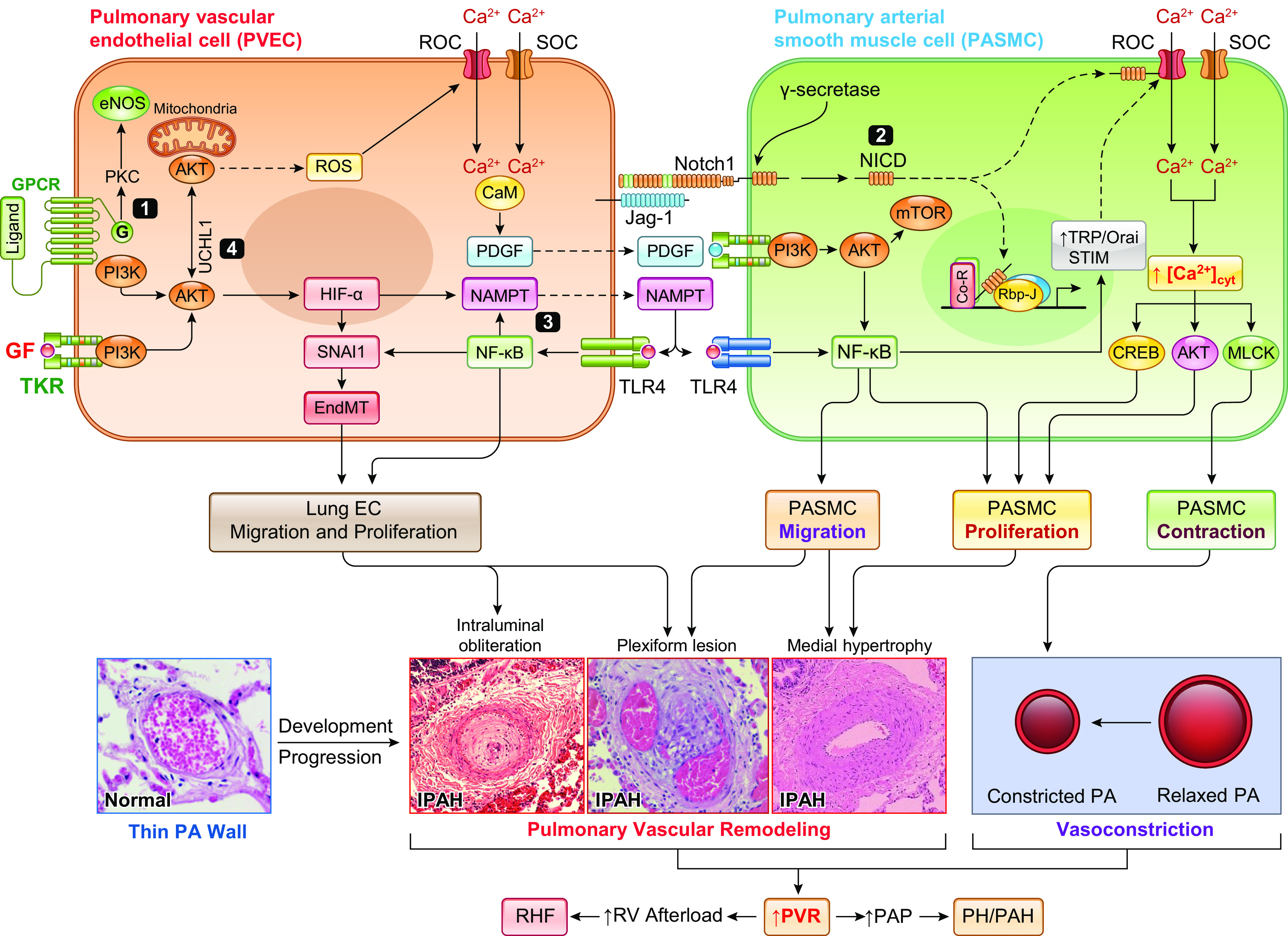
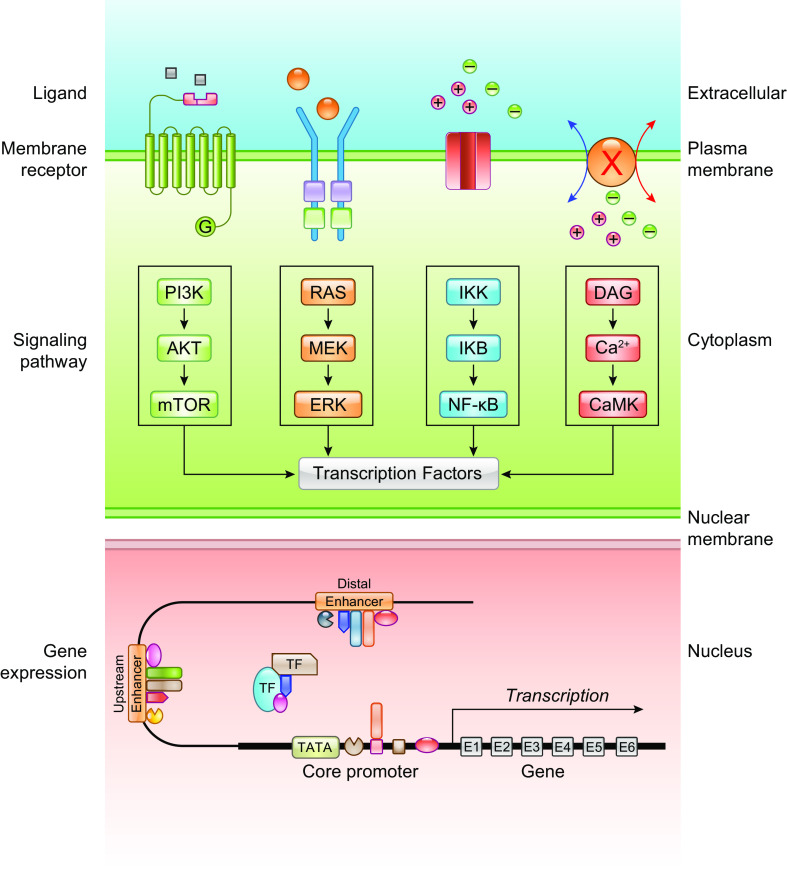
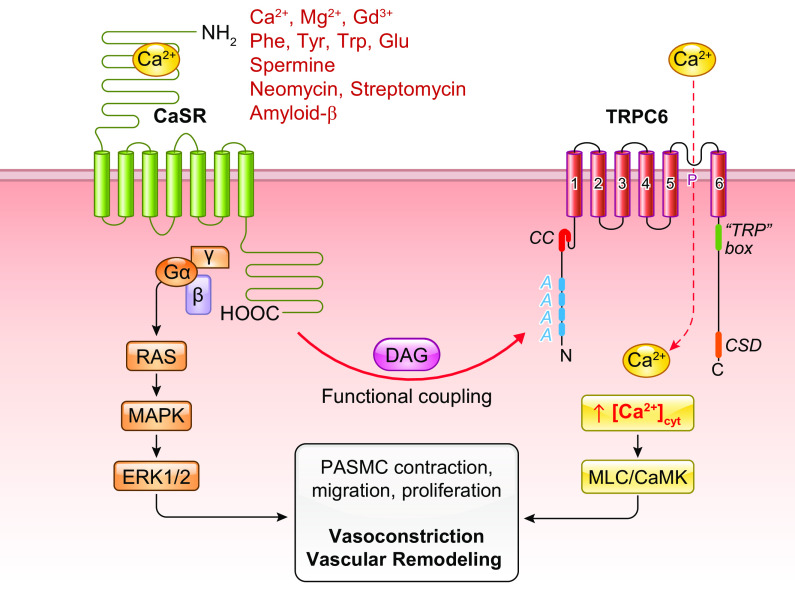
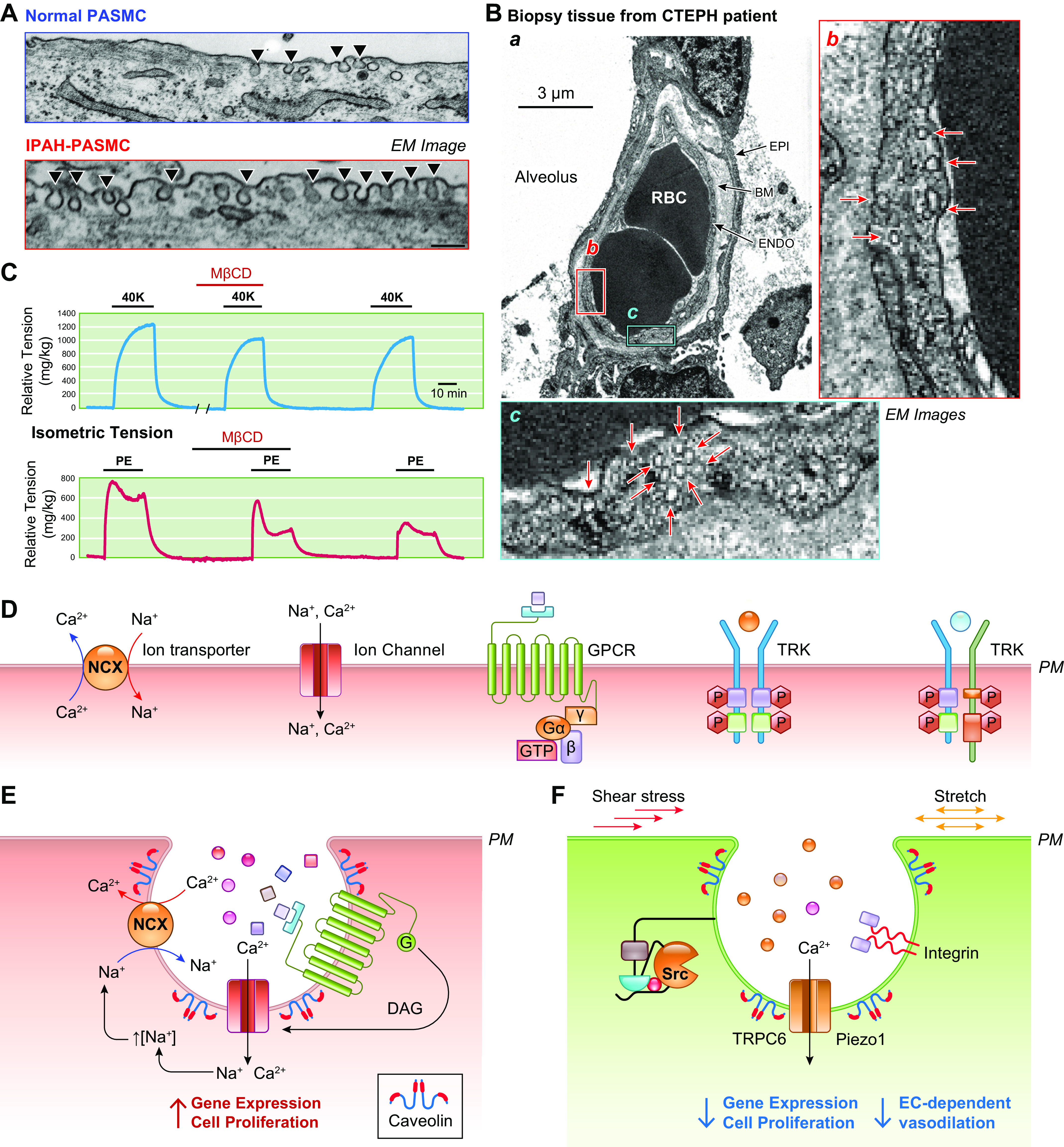
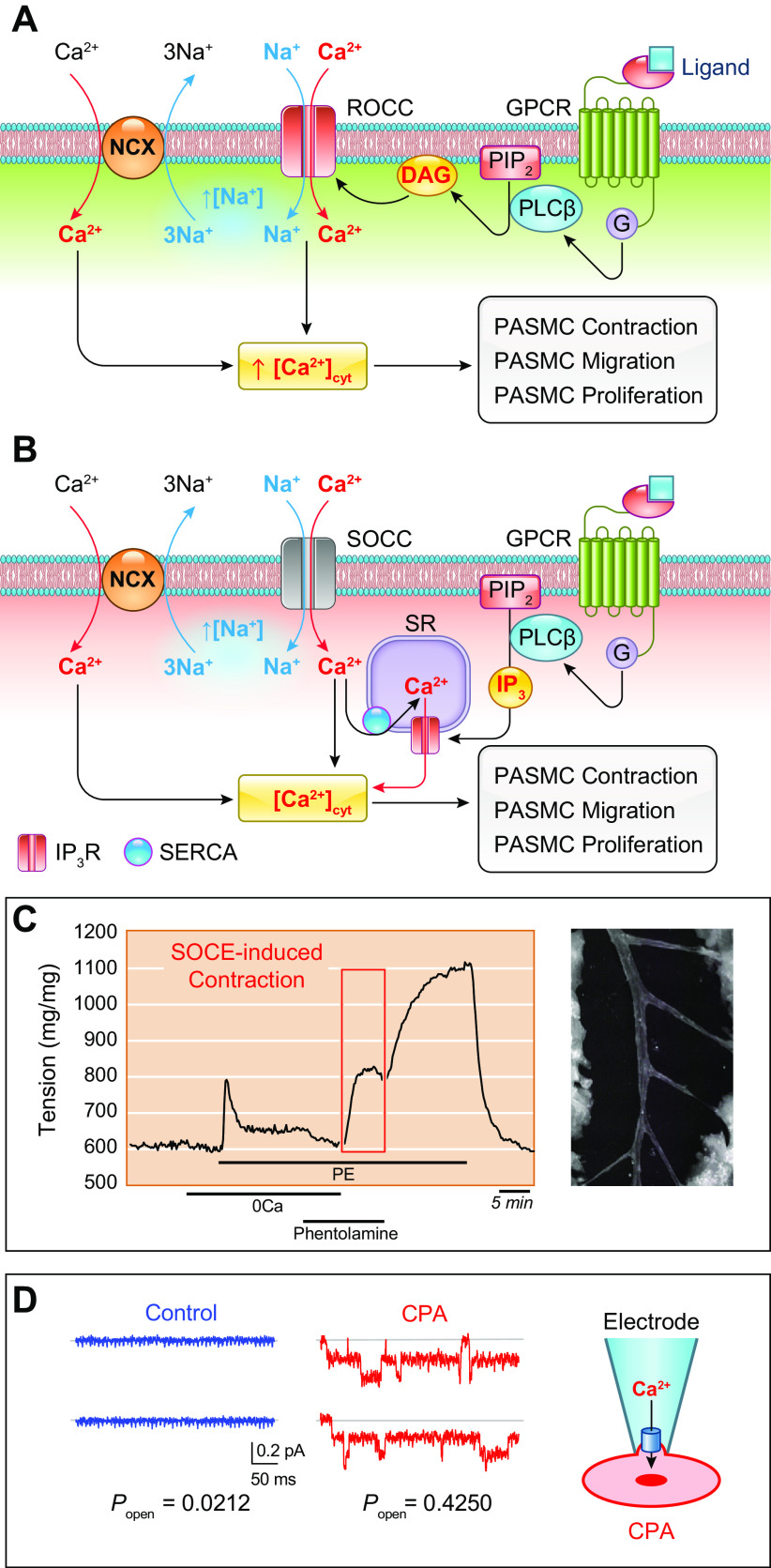
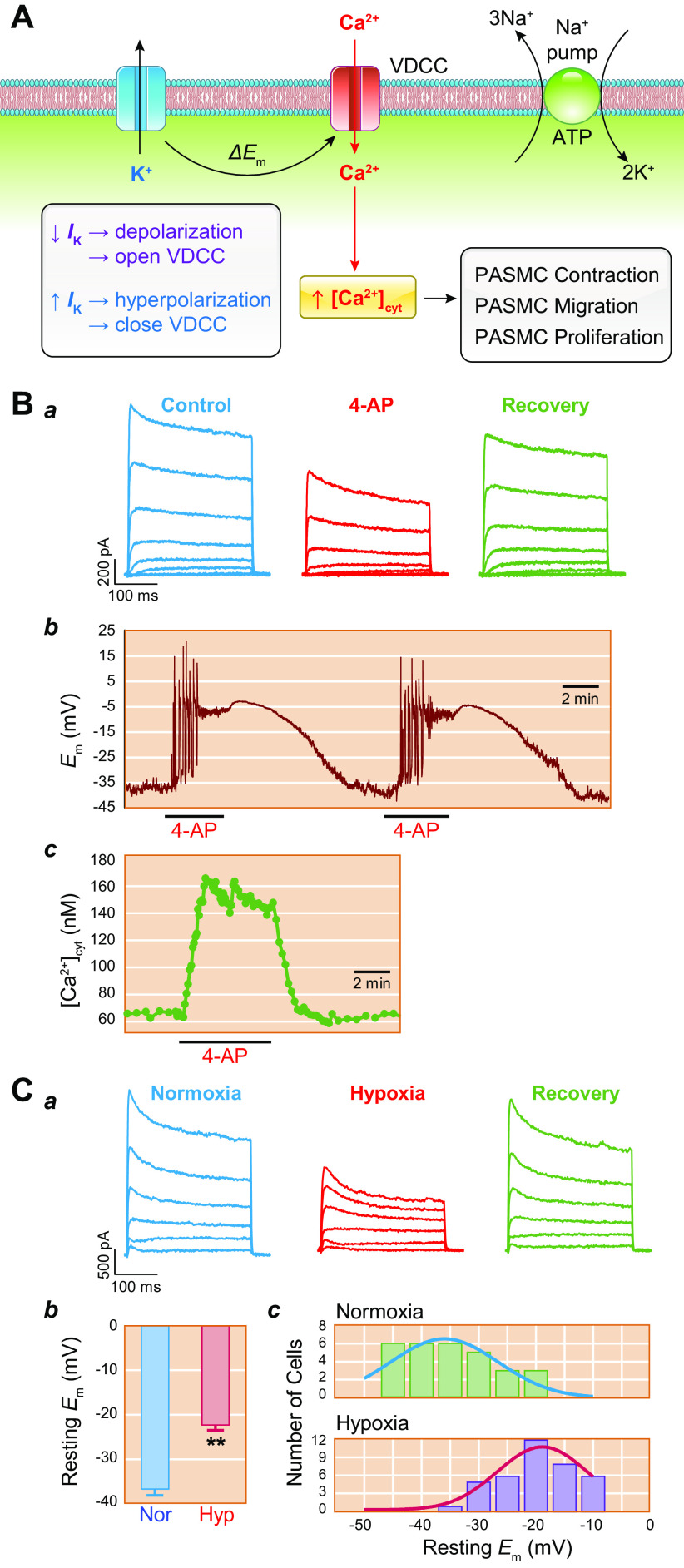
The pulmonary circulation is a low-resistance, low-pressure, and high-compliance system that allows the lungs to receive the entire cardiac output. Pulmonary arterial pressure is a function of cardiac output and pulmonary vascular resistance, and pulmonary vascular resistance is inversely proportional to the fourth power of the intraluminal radius of the pulmonary artery. Therefore, a very small decrease of the pulmonary vascular lumen diameter results in a significant increase in pulmonary vascular resistance and pulmonary arterial pressure. Pulmonary arterial hypertension is a fatal and progressive disease with poor prognosis. Regardless of the initial pathogenic triggers, sustained pulmonary vasoconstriction, concentric vascular remodeling, occlusive intimal lesions, in situ thrombosis, and vascular wall stiffening are the major and direct causes for elevated pulmonary vascular resistance in patients with pulmonary arterial hypertension and other forms of precapillary pulmonary hypertension. In this review, we aim to discuss the basic principles and physiological mechanisms involved in the regulation of lung vascular hemodynamics and pulmonary vascular function, the changes in the pulmonary vasculature that contribute to the increased vascular resistance and arterial pressure, and the pathogenic mechanisms involved in the development and progression of pulmonary hypertension. We focus on reviewing the pathogenic roles of membrane receptors, ion channels, and intracellular Ca2+ signaling in pulmonary vascular smooth muscle cells in the development and progression of pulmonary hypertension.
Keywords: Ca2+ signaling; ion channel; membrane receptor; pulmonary arterial hypertension; pulmonary circulation.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
References
-
- Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RM, Brida M, Carlsen J, Coats AJ, Escribano-Subias P, Ferrari P, Ferreira DS, Ghofrani HA, Giannakoulas G, Kiely DG, Mayer E, Meszaros G, Nagavci B, Olsson KM, Pepke-Zaba J, Quint JK, Rådegran G, Simonneau G, Sitbon O, Tonia T, Toshner M, Vachiery JL, Vonk Noordegraaf A, Delcroix M, Rosenkranz S; the ESC/ERS Scientific Document Group. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 61: 2200879, 2023. doi: 10.1183/13993003.00879-2022. - DOI - PubMed
-
- Remillard CY. Characterization of hemodynamics in patients with idiopathic and thromboembolic pulmonary hypertension. Clin Med Insights Circ Respir Pulm Med 2: 59–68, 2008. doi: 10.4137/CCRPM.S696. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous