

The Role of Copper Homeostasis in Brain Disease
- PMID: 36430330
- PMCID: PMC9698384
- DOI: 10.3390/ijms232213850
The Role of Copper Homeostasis in Brain Disease
Abstract
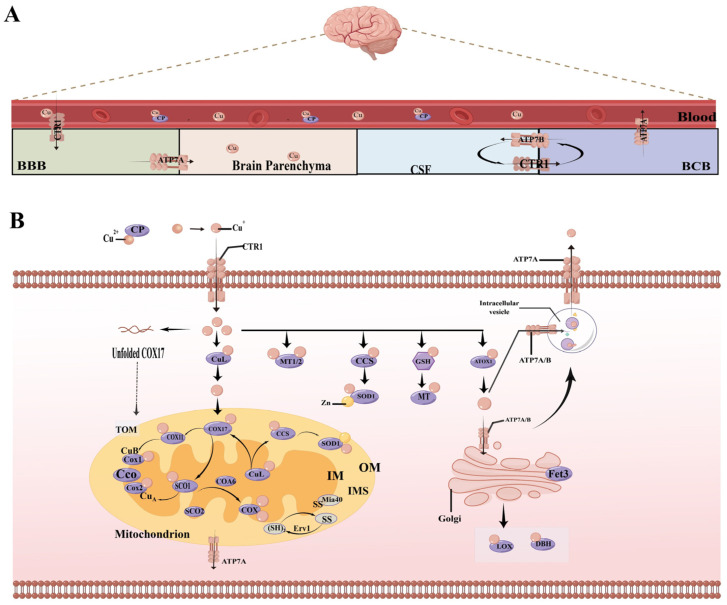
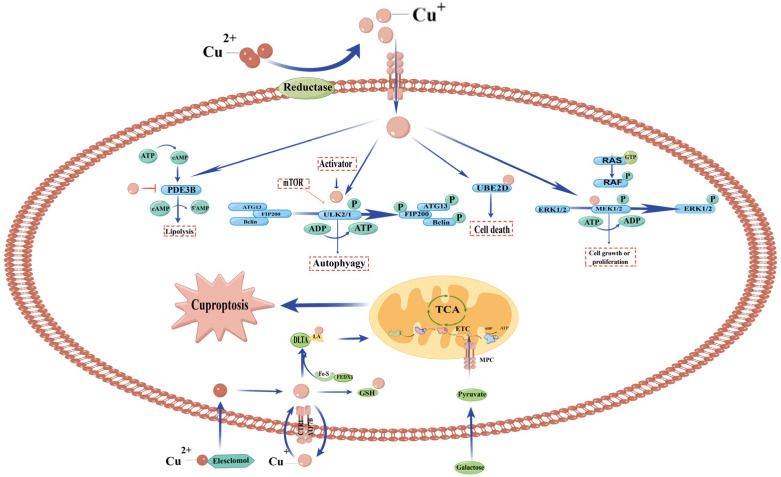
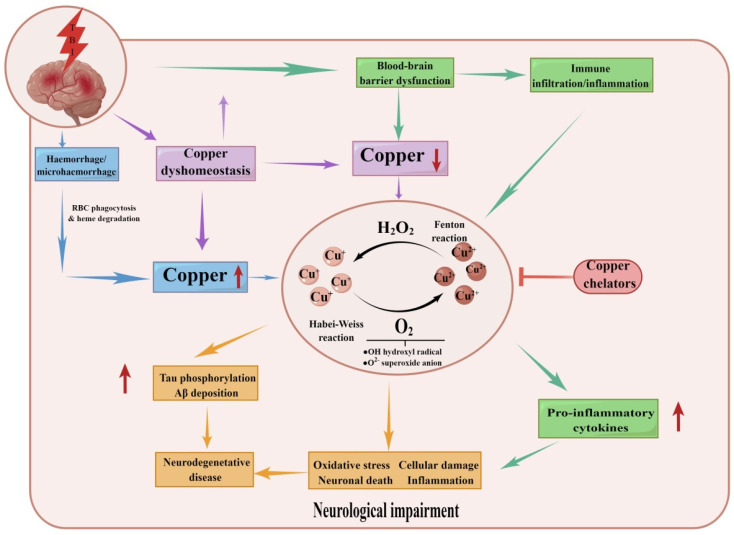
In the human body, copper is an important trace element and is a cofactor for several important enzymes involved in energy production, iron metabolism, neuropeptide activation, connective tissue synthesis, and neurotransmitter synthesis. Copper is also necessary for cellular processes, such as the regulation of intracellular signal transduction, catecholamine balance, myelination of neurons, and efficient synaptic transmission in the central nervous system. Copper is naturally present in some foods and is available as a dietary supplement. Only small amounts of copper are typically stored in the body and a large amount of copper is excreted through bile and urine. Given the critical role of copper in a breadth of cellular processes, local concentrations of copper and the cellular distribution of copper transporter proteins in the brain are important to maintain the steady state of the internal environment. The dysfunction of copper metabolism or regulatory pathways results in an imbalance in copper homeostasis in the brain, which can lead to a myriad of acute and chronic pathological effects on neurological function. It suggests a unique mechanism linking copper homeostasis and neuronal activation within the central nervous system. This article explores the relationship between impaired copper homeostasis and neuropathophysiological progress in brain diseases.
Keywords: brain injury; cognition; copper; cuproptosis; neurodegeneration.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- No. 82071382, No. 81601306/National Natural Science Foundation of China
- PAPD/The Priority Academic Program Development of Jiangsu Higher Education Institutions
- F202013/The Jiangsu Maternal and Child Health Research Key Project
- 2022/Jiangsu 333 High Level Talent Training Project
- QNRC2016245/Jiangsu Talent Youth Medical Program
LinkOut - more resources
Full Text Sources
Medical
