

Immune phenotypes that are associated with subsequent COVID-19 severity inferred from post-recovery samples
- PMID: 36433939
- PMCID: PMC9700777
- DOI: 10.1038/s41467-022-34638-2
Immune phenotypes that are associated with subsequent COVID-19 severity inferred from post-recovery samples
Abstract
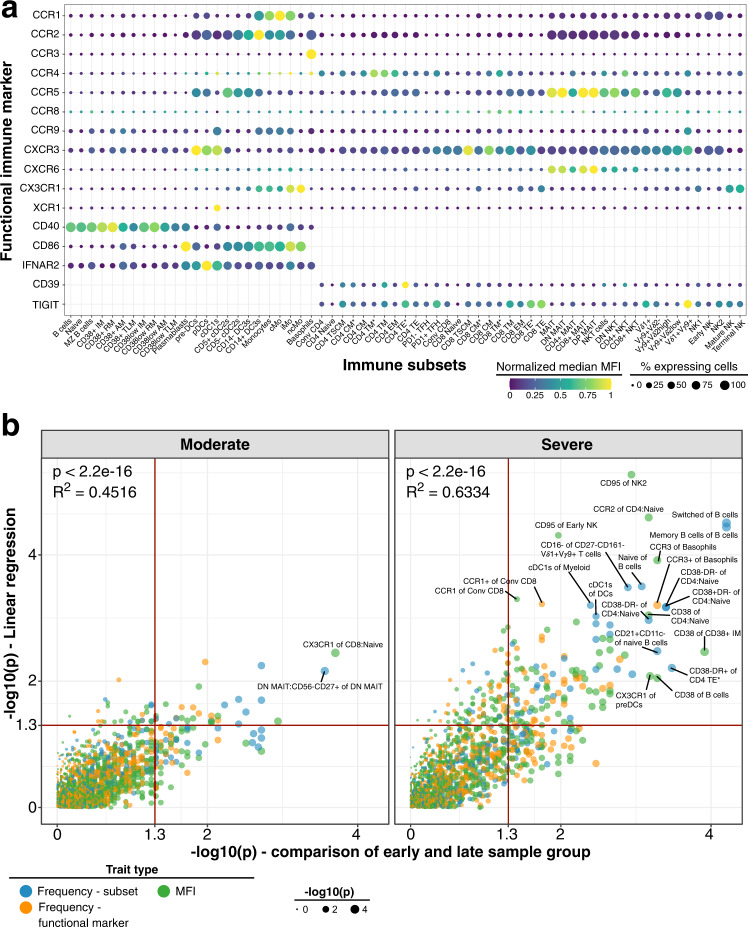
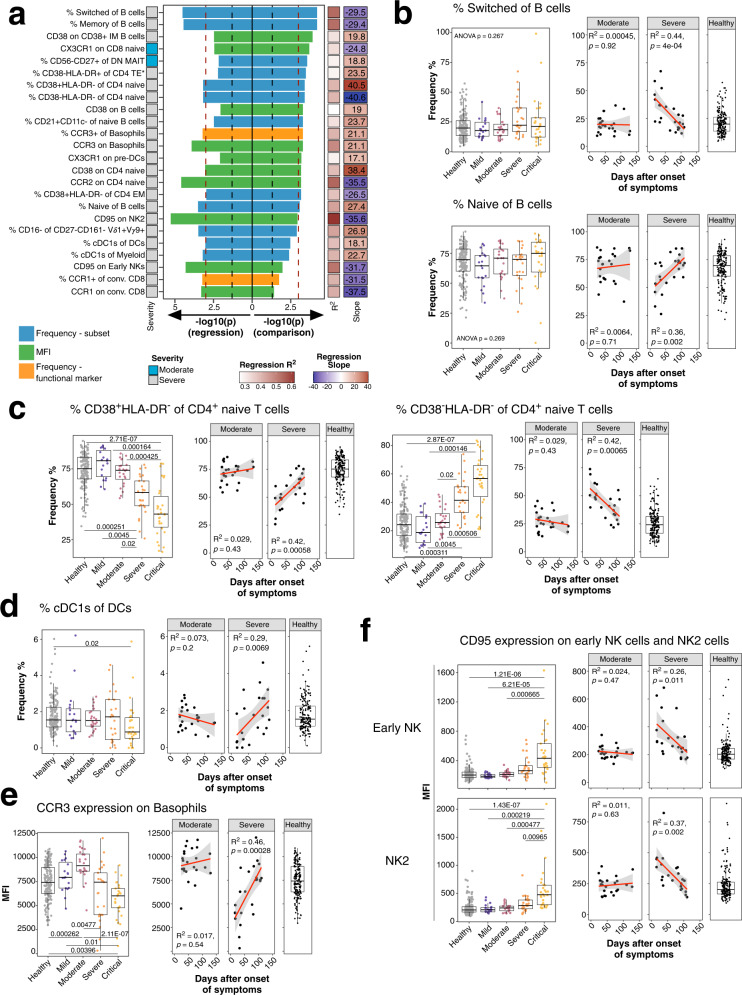
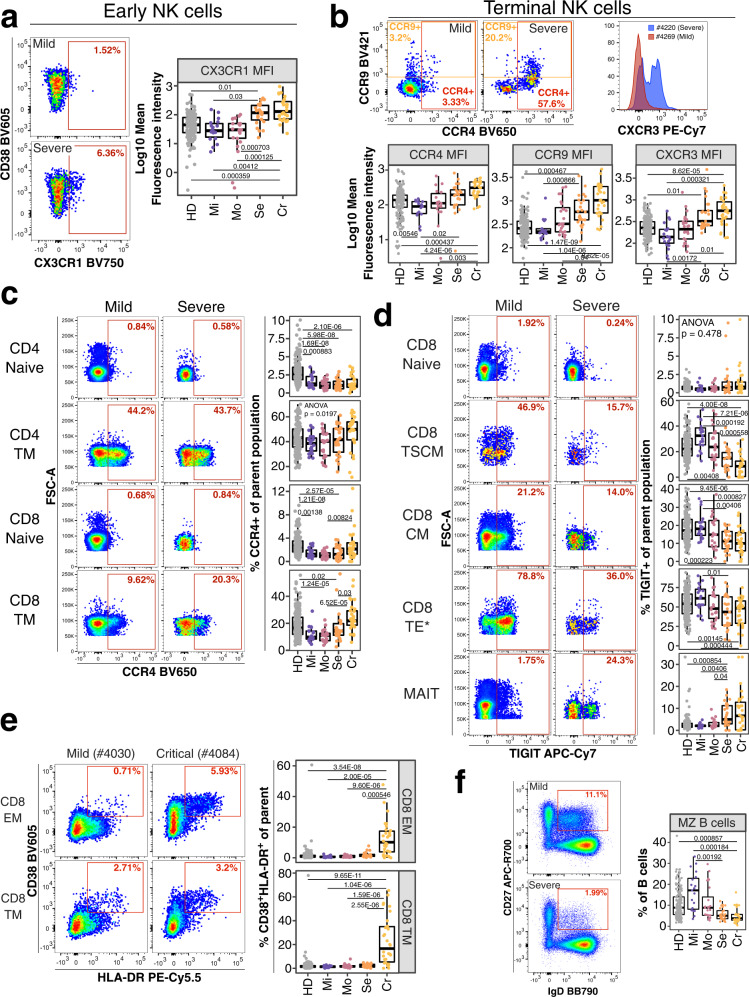
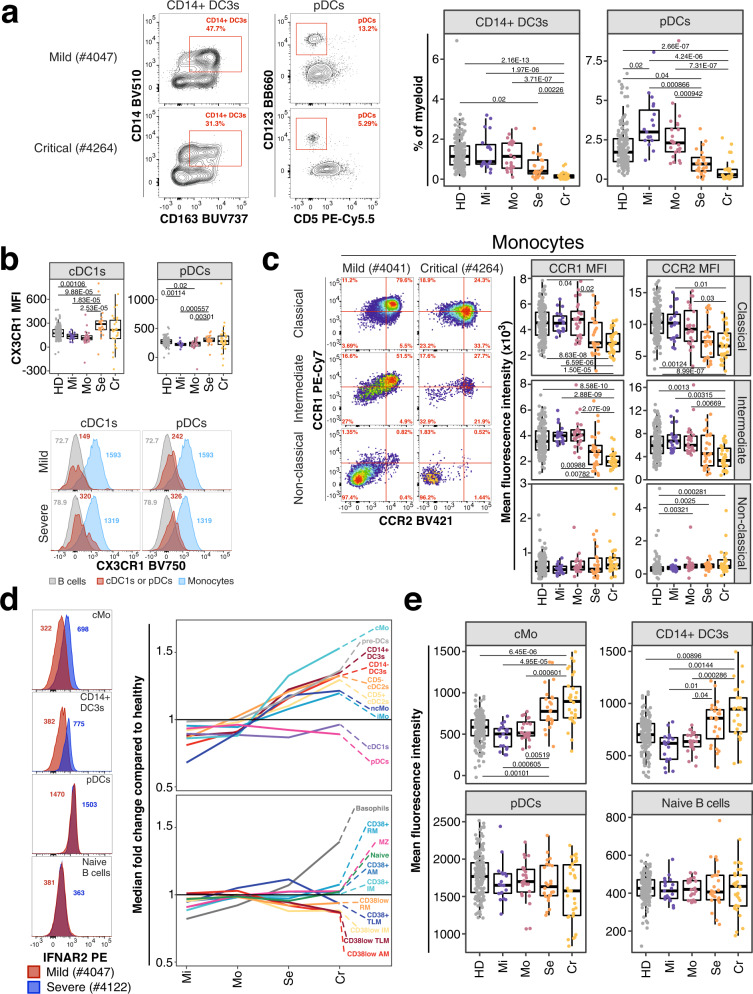
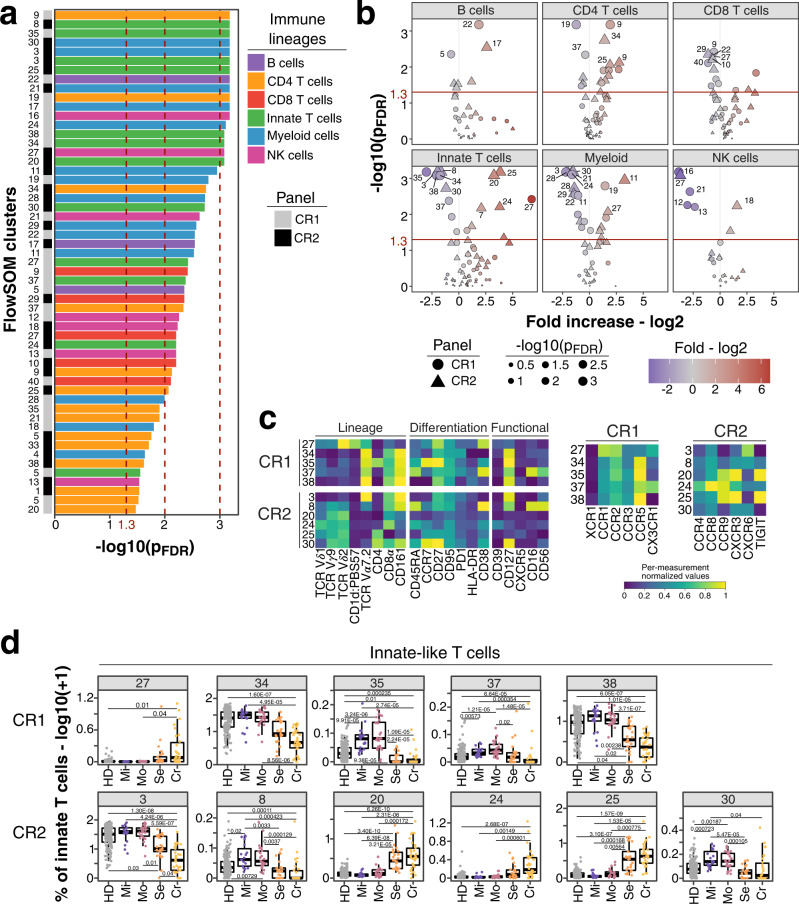
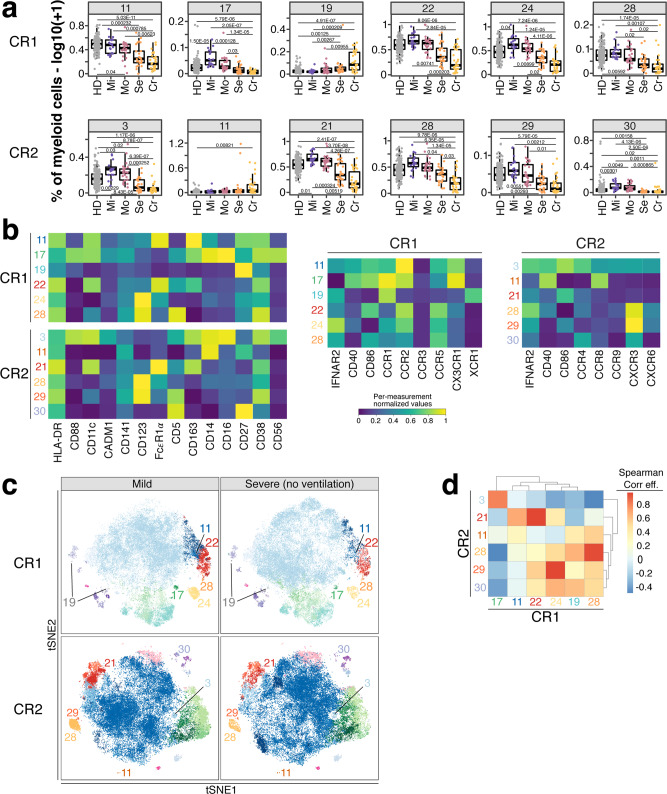
Severe COVID-19 causes profound immune perturbations, but pre-infection immune signatures contributing to severe COVID-19 remain unknown. Genome-wide association studies (GWAS) identified strong associations between severe disease and several chemokine receptors and molecules from the type I interferon pathway. Here, we define immune signatures associated with severe COVID-19 using high-dimensional flow cytometry. We measure the cells of the peripheral immune system from individuals who recovered from mild, moderate, severe or critical COVID-19 and focused only on those immune signatures returning to steady-state. Individuals that suffered from severe COVID-19 show reduced frequencies of T cell, mucosal-associated invariant T cell (MAIT) and dendritic cell (DC) subsets and altered chemokine receptor expression on several subsets, such as reduced levels of CCR1 and CCR2 on monocyte subsets. Furthermore, we find reduced frequencies of type I interferon-producing plasmacytoid DCs and altered IFNAR2 expression on several myeloid cells in individuals recovered from severe COVID-19. Thus, these data identify potential immune mechanisms contributing to severe COVID-19.
© 2022. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
Immune phenotypes that predict COVID-19 severity.Res Sq [Preprint]. 2022 Mar 10:rs.3.rs-1378671. doi: 10.21203/rs.3.rs-1378671/v1. Res Sq. 2022. Update in: Nat Commun. 2022 Nov 25;13(1):7255. doi: 10.1038/s41467-022-34638-2. PMID: 35291290 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
