

Lynch syndrome, molecular mechanisms and variant classification
- PMID: 36434153
- PMCID: PMC9978028
- DOI: 10.1038/s41416-022-02059-z
Lynch syndrome, molecular mechanisms and variant classification
Abstract
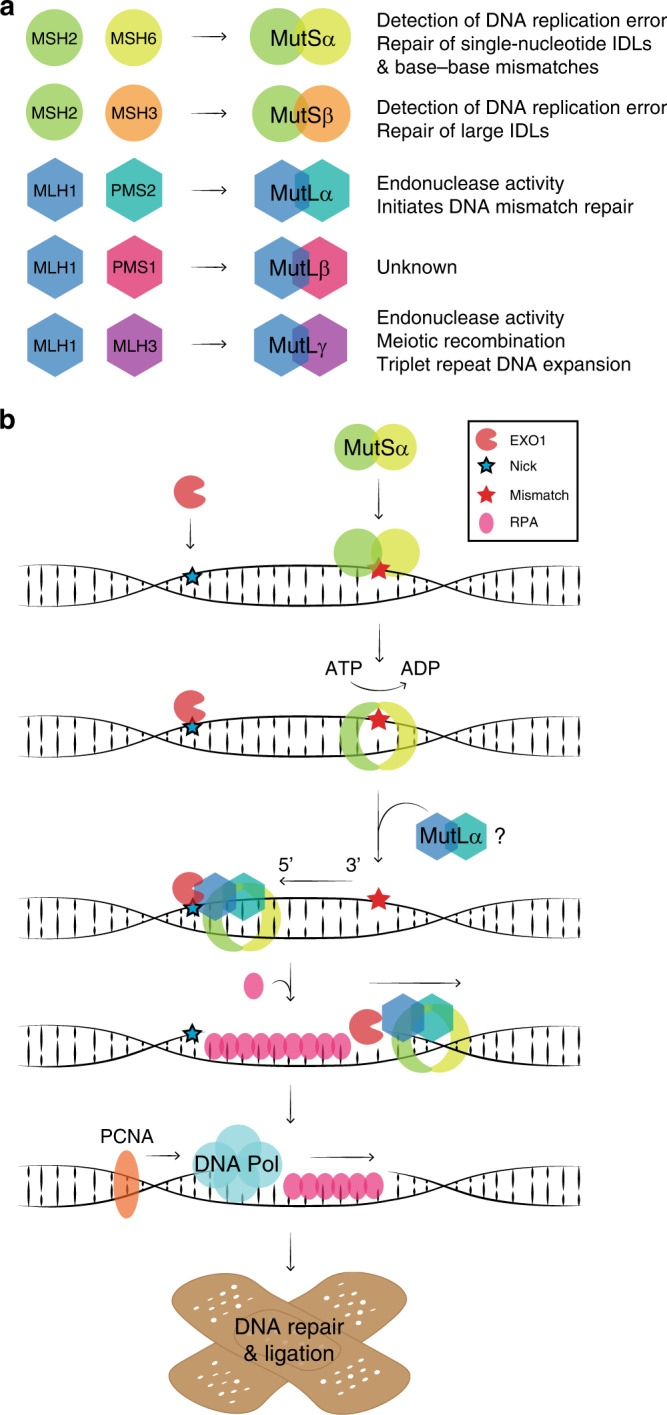
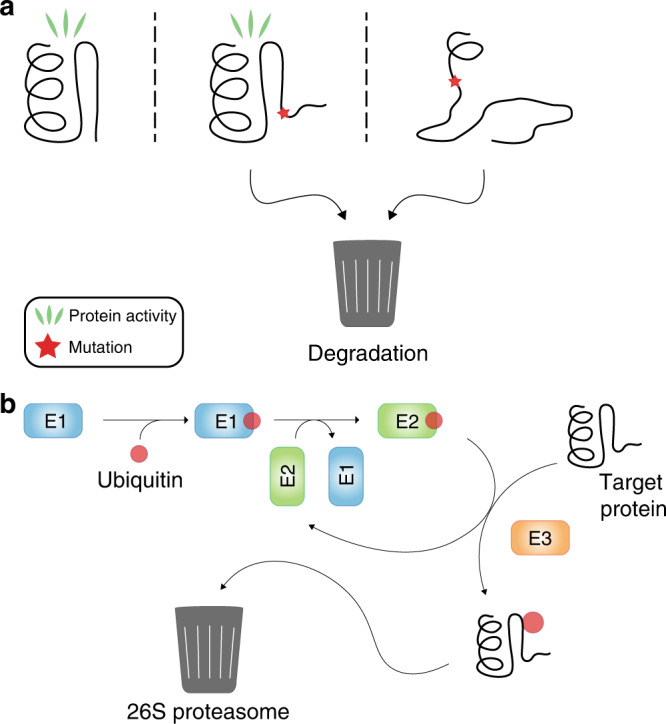
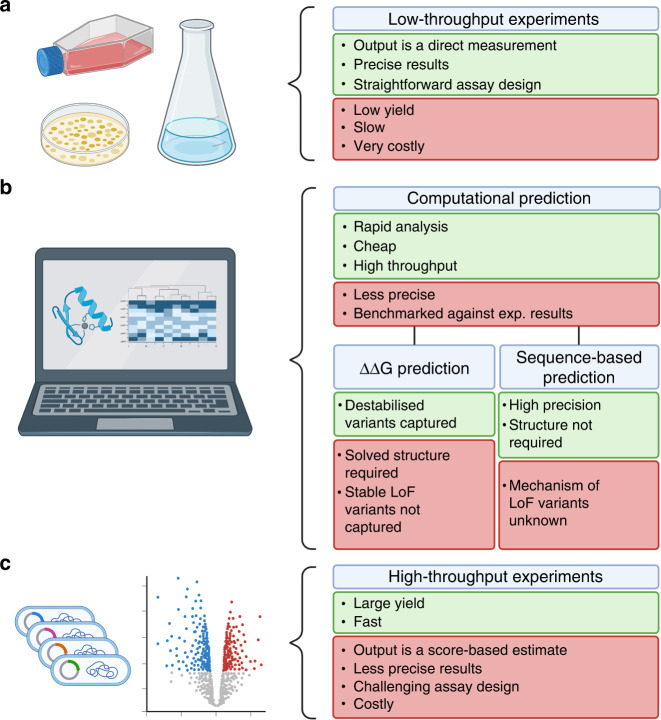
Patients with the heritable cancer disease, Lynch syndrome, carry germline variants in the MLH1, MSH2, MSH6 and PMS2 genes, encoding the central components of the DNA mismatch repair system. Loss-of-function variants disrupt the DNA mismatch repair system and give rise to a detrimental increase in the cellular mutational burden and cancer development. The treatment prospects for Lynch syndrome rely heavily on early diagnosis; however, accurate diagnosis is inextricably linked to correct clinical interpretation of individual variants. Protein variant classification traditionally relies on cumulative information from occurrence in patients, as well as experimental testing of the individual variants. The complexity of variant classification is due to (1) that variants of unknown significance are rare in the population and phenotypic information on the specific variants is missing, and (2) that individual variant testing is challenging, costly and slow. Here, we summarise recent developments in high-throughput technologies and computational prediction tools for the assessment of variants of unknown significance in Lynch syndrome. These approaches may vastly increase the number of interpretable variants and could also provide important mechanistic insights into the disease. These insights may in turn pave the road towards developing personalised treatment approaches for Lynch syndrome.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Dominguez-Valentin M, Sampson JR, Seppälä TT, ten Broeke SW, Plazzer J-P, Nakken S, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med. 2020;22:15–25. doi: 10.1038/s41436-019-0596-9. - DOI - PMC - PubMed
-
- Møller P, Seppälä T, Bernstein I, Holinski-Feder E, Sala P, Evans DG, et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut. 2017;66:464–72. doi: 10.1136/gutjnl-2015-309675. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
