

Mutual regulation between phosphofructokinase 1 platelet isoform and VEGF promotes glioblastoma tumor growth
- PMID: 36435833
- PMCID: PMC9701207
- DOI: 10.1038/s41419-022-05449-6
Mutual regulation between phosphofructokinase 1 platelet isoform and VEGF promotes glioblastoma tumor growth
Abstract
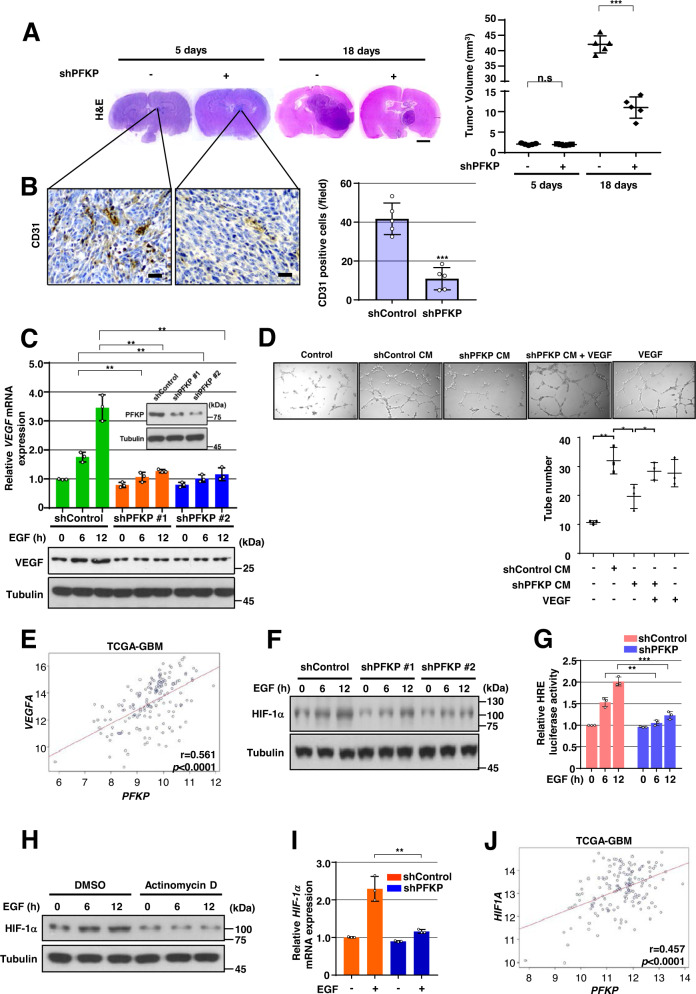
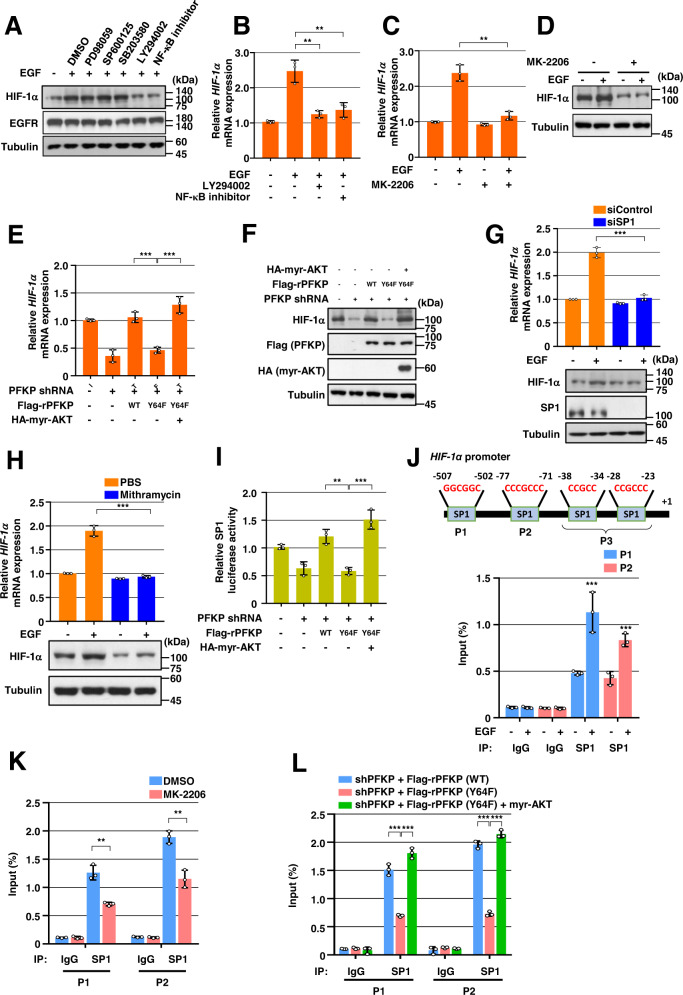
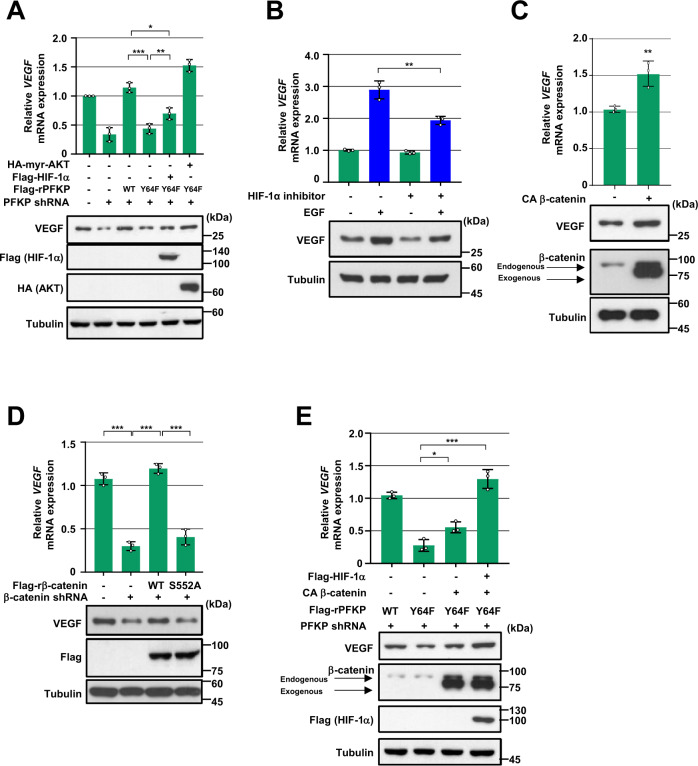
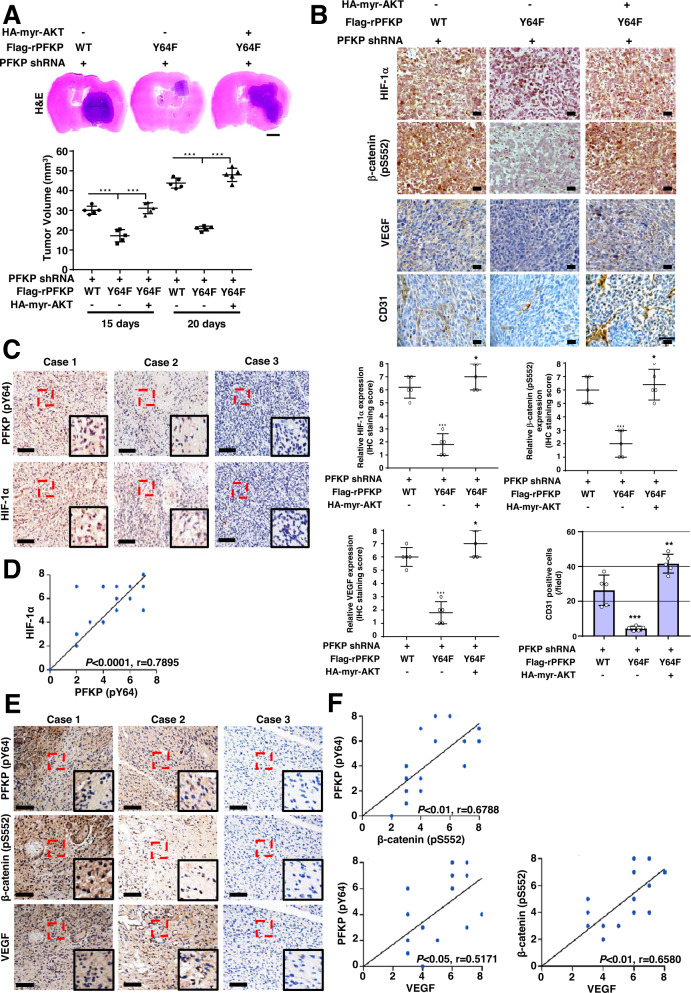
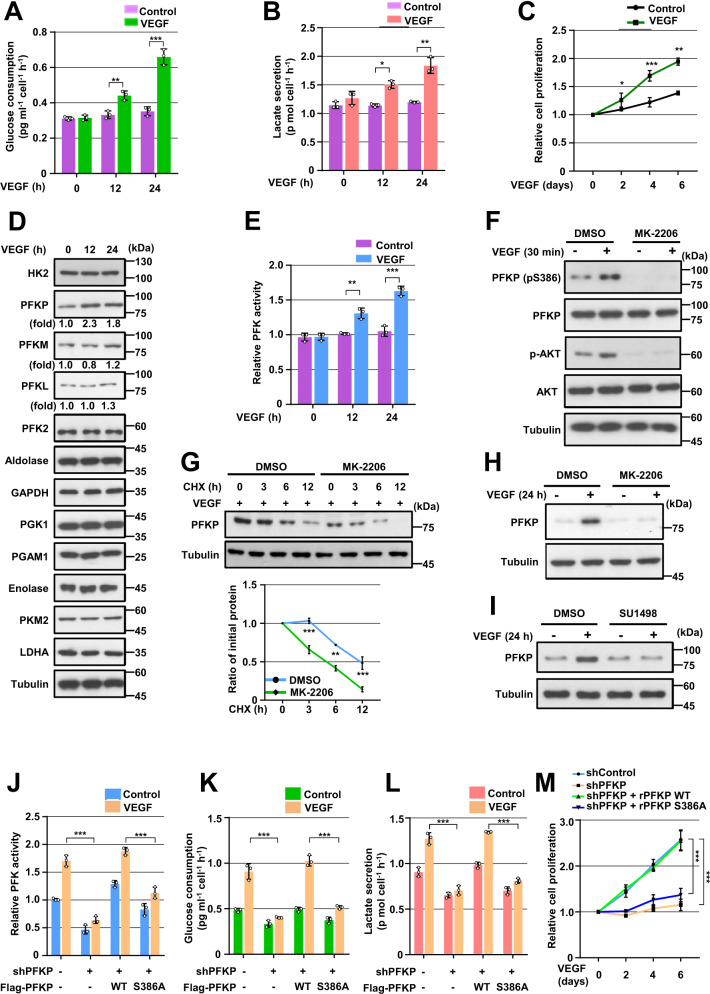
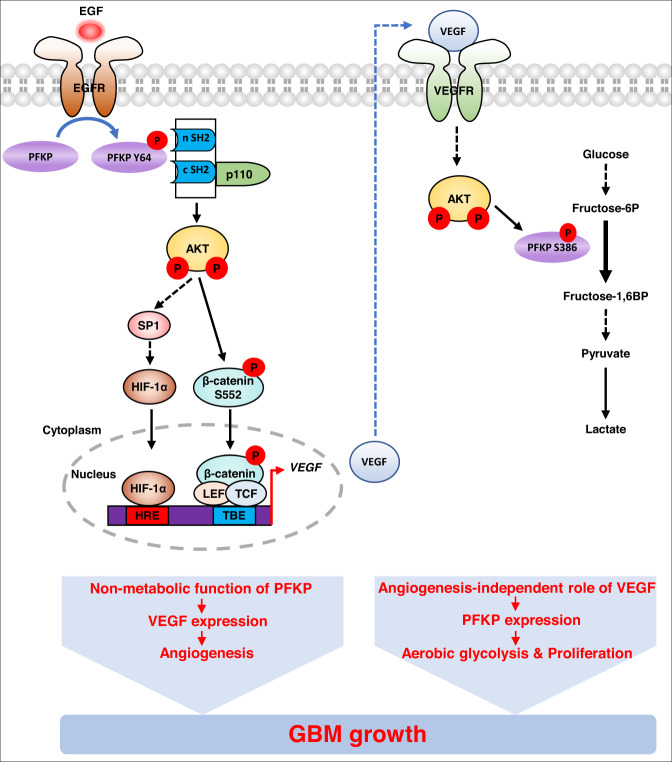
Glioblastoma (GBM) is a highly vascular malignant brain tumor that overexpresses vascular endothelial growth factor (VEGF) and phosphofructokinase 1 platelet isoform (PFKP), which catalyzes a rate-limiting reaction in glycolysis. However, whether PFKP and VEGF are reciprocally regulated during GBM tumor growth remains unknown. Here, we show that PFKP can promote EGFR activation-induced VEGF expression in HIF-1α-dependent and -independent manners in GBM cells. Importantly, we demonstrate that EGFR-phosphorylated PFKP Y64 has critical roles in both AKT/SP1-mediated transcriptional expression of HIF-1α and in the AKT-mediated β-catenin S552 phosphorylation, to fully enhance VEGF transcription, subsequently promoting blood vessel formation and brain tumor growth. Levels of PFKP Y64 phosphorylation in human GBM specimens are positively correlated with HIF-1α expression, β-catenin S552 phosphorylation, and VEGF expression. Conversely, VEGF upregulates PFKP expression in a PFKP S386 phosphorylation-dependent manner, leading to increased PFK enzyme activity, aerobic glycolysis, and proliferation in GBM cells. These findings highlight a novel mechanism underlying the mutual regulation that occurs between PFKP and VEGF for promoting GBM tumor growth and also suggest that targeting the PFKP/VEGF regulatory loop might show therapeutic potential for treating GBM patients.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
