

ARID1A loss induces polymorphonuclear myeloid-derived suppressor cell chemotaxis and promotes prostate cancer progression
- PMID: 36435834
- PMCID: PMC9701216
- DOI: 10.1038/s41467-022-34871-9
ARID1A loss induces polymorphonuclear myeloid-derived suppressor cell chemotaxis and promotes prostate cancer progression
Abstract
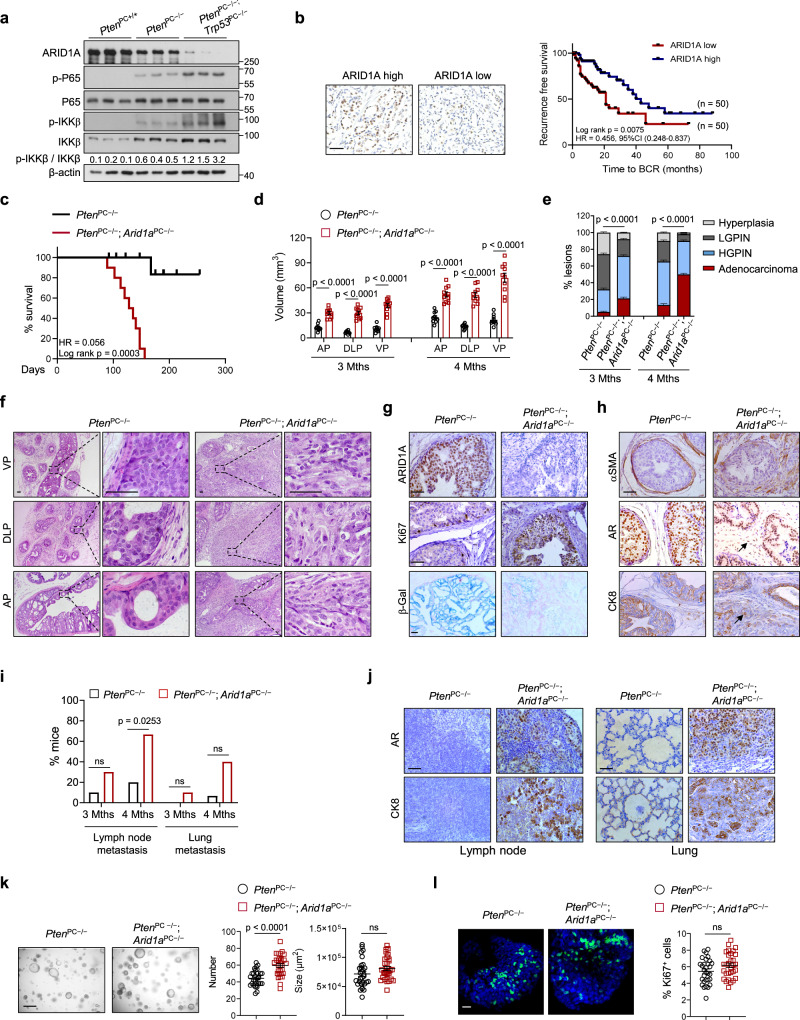
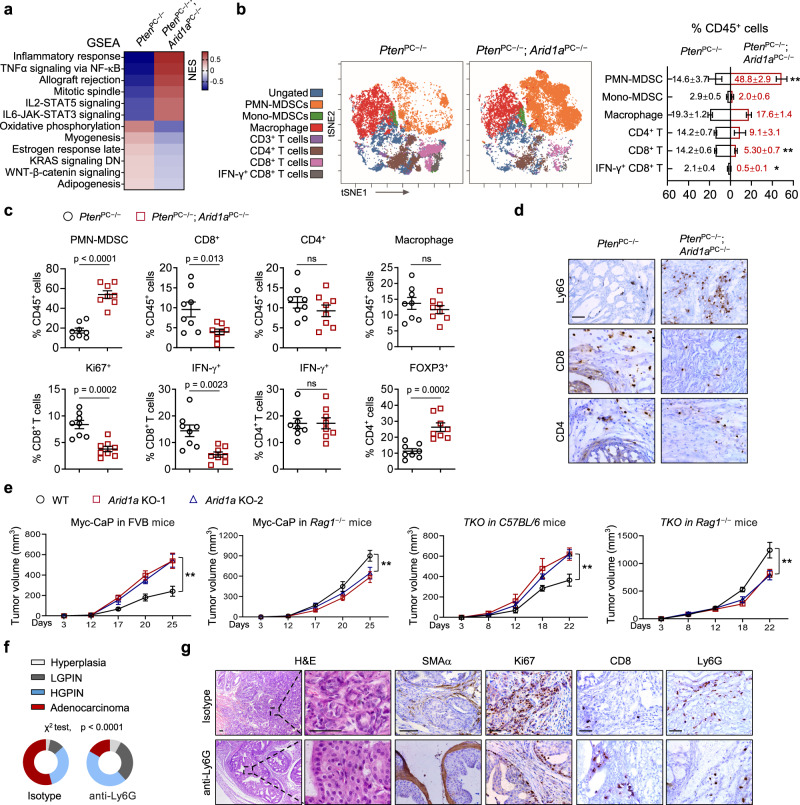
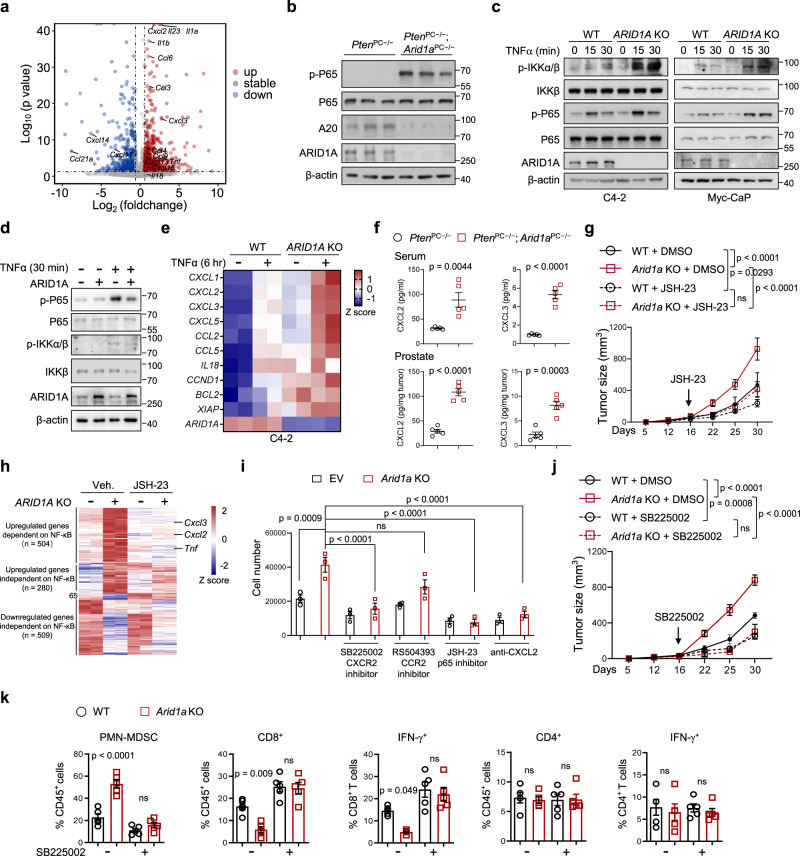
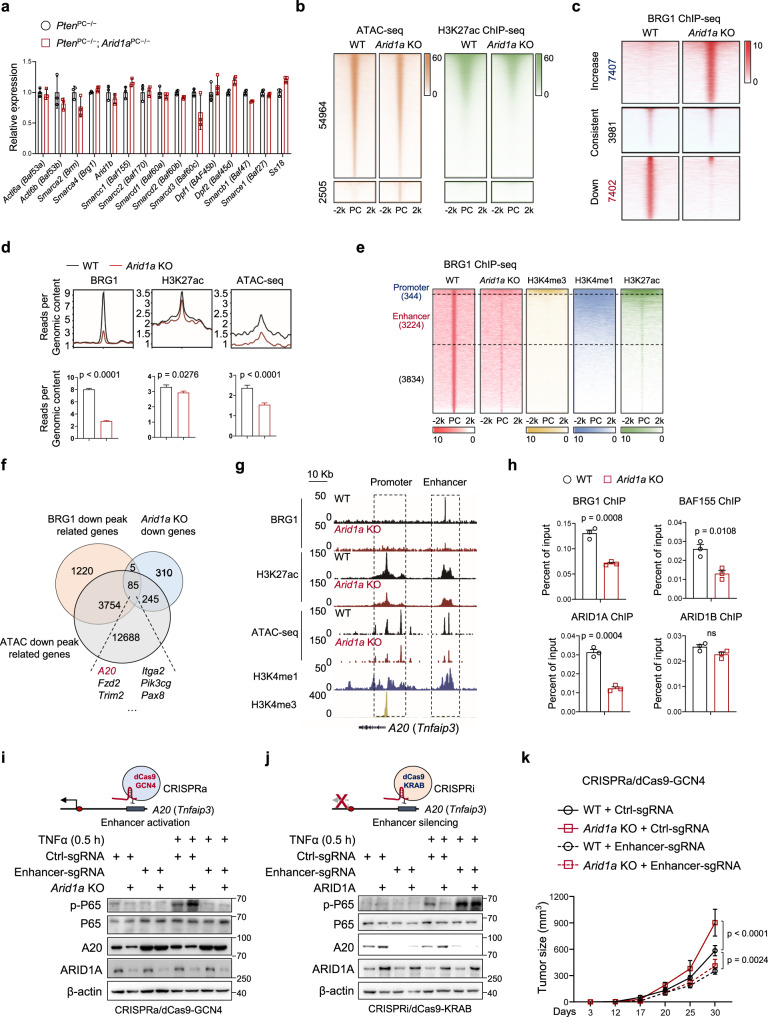
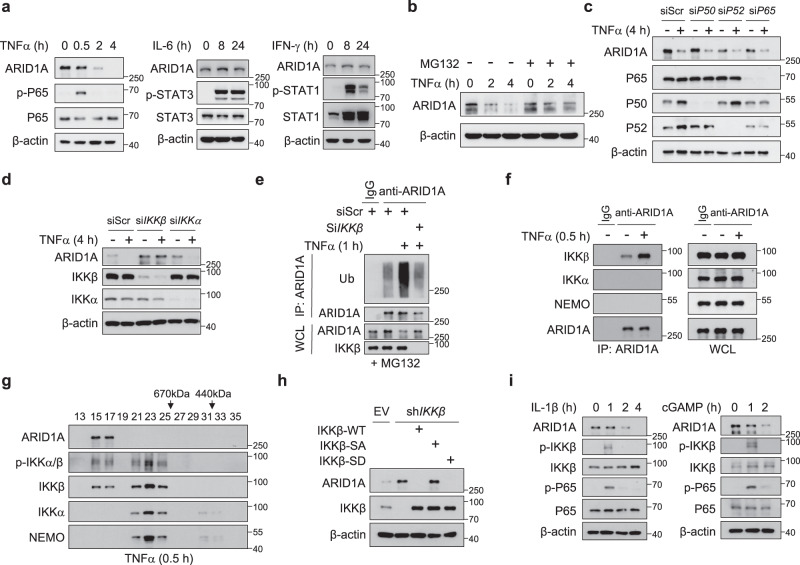
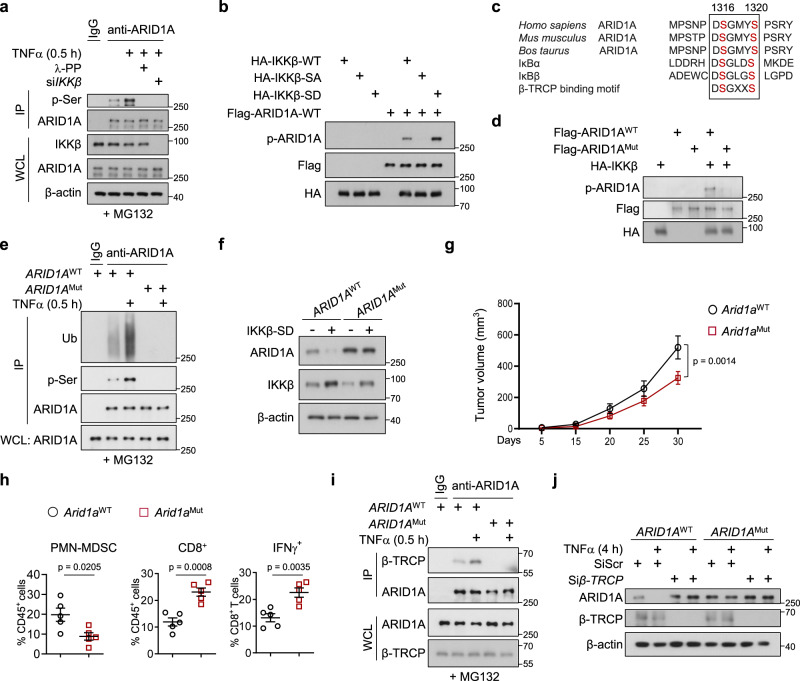
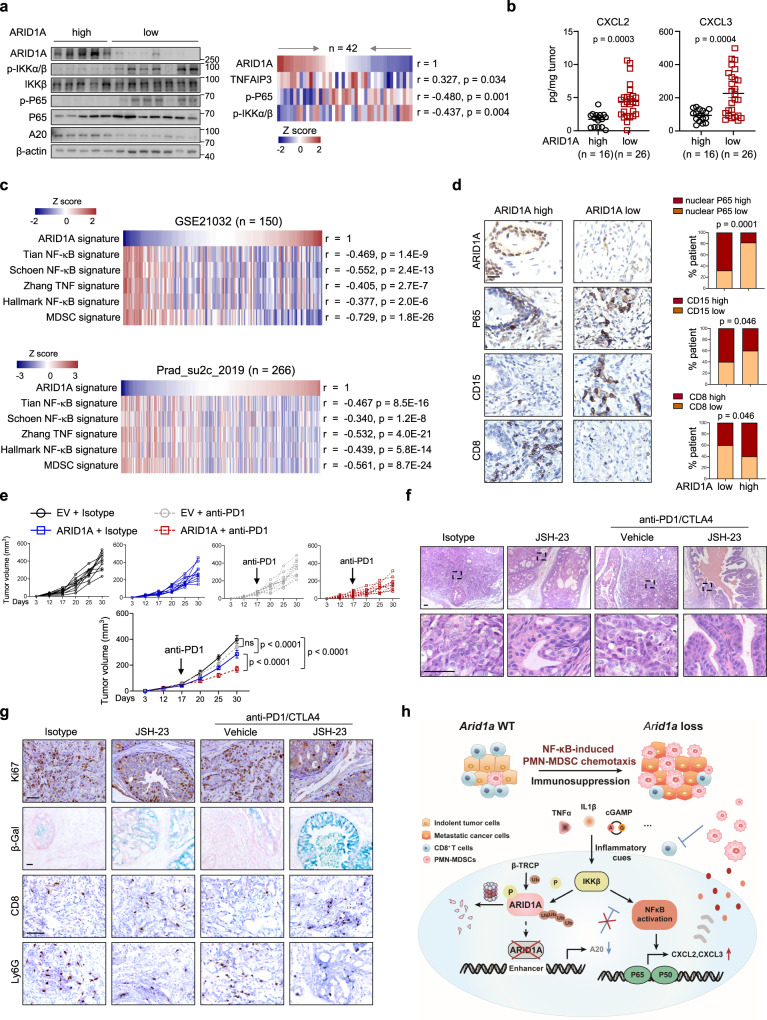
Chronic inflammation and an immunosuppressive microenvironment promote prostate cancer (PCa) progression and diminish the response to immune checkpoint blockade (ICB) therapies. However, it remains unclear how and to what extent these two events are coordinated. Here, we show that ARID1A, a subunit of the SWI/SNF chromatin remodeling complex, functions downstream of inflammation-induced IKKβ activation to shape the immunosuppressive tumor microenvironment (TME). Prostate-specific deletion of Arid1a cooperates with Pten loss to accelerate prostate tumorigenesis. We identify polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) as the major infiltrating immune cell type that causes immune evasion and reveal that neutralization of PMN-MDSCs restricts the progression of Arid1a-deficient tumors. Mechanistically, inflammatory cues activate IKKβ to phosphorylate ARID1A, leading to its degradation via β-TRCP. ARID1A downregulation in turn silences the enhancer of A20 deubiquitinase, a critical negative regulator of NF-κB signaling, and thereby unleashes CXCR2 ligand-mediated MDSC chemotaxis. Importantly, our results support the therapeutic strategy of anti-NF-κB antibody or targeting CXCR2 combined with ICB for advanced PCa. Together, our findings highlight that the IKKβ/ARID1A/NF-κB feedback axis integrates inflammation and immunosuppression to promote PCa progression.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
