

Expanding known viral diversity in plants: virome of 161 species alongside an ancient canal
- PMID: 36437477
- PMCID: PMC9703751
- DOI: 10.1186/s40793-022-00453-x
Expanding known viral diversity in plants: virome of 161 species alongside an ancient canal
Abstract
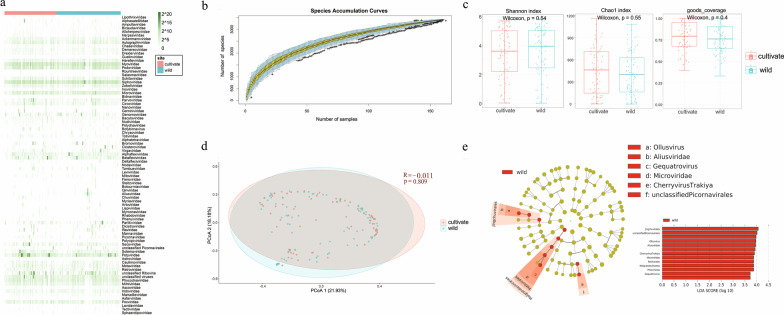
Background: Since viral metagenomic approach was applied to discover plant viruses for the first time in 2006, many plant viruses had been identified from cultivated and non-cultivated plants. These previous researches exposed that the viral communities (virome) of plants have still largely uncharacterized. Here, we investigated the virome in 161 species belonging to 38 plant orders found in a riverside ecosystem.
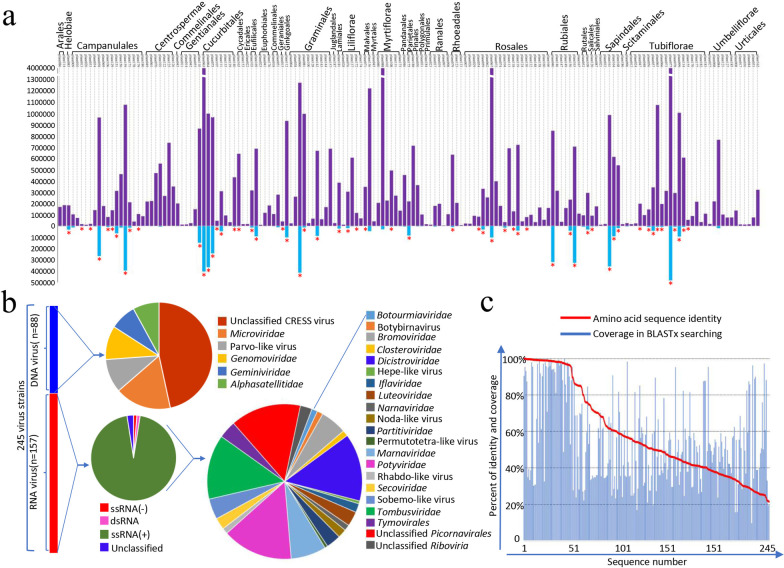
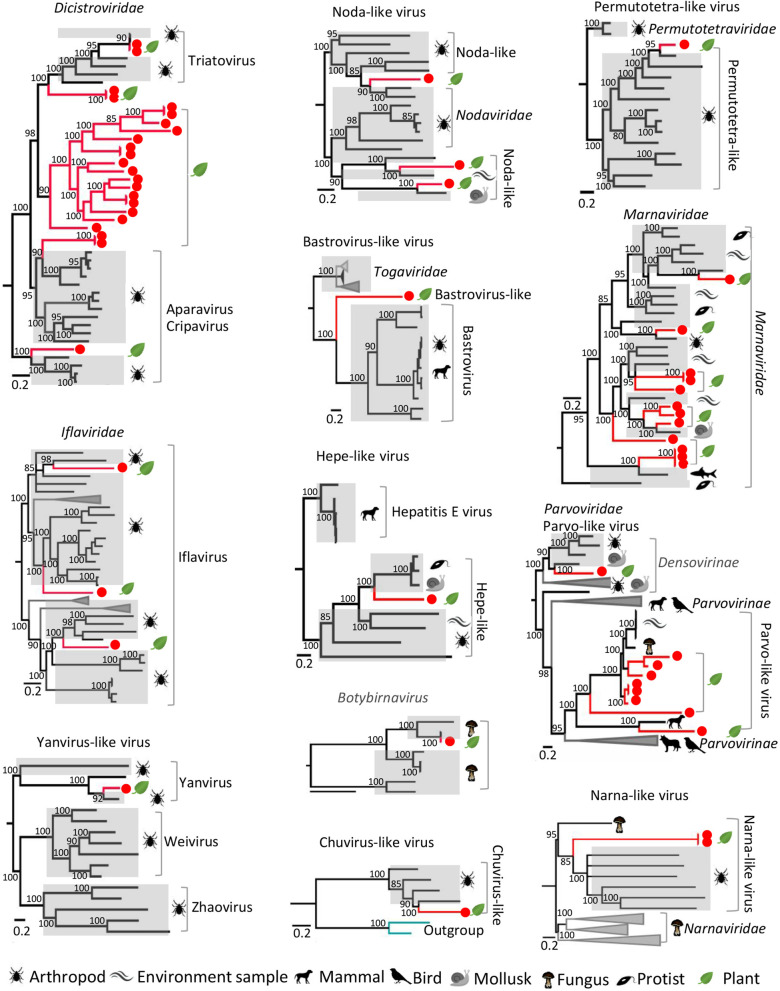
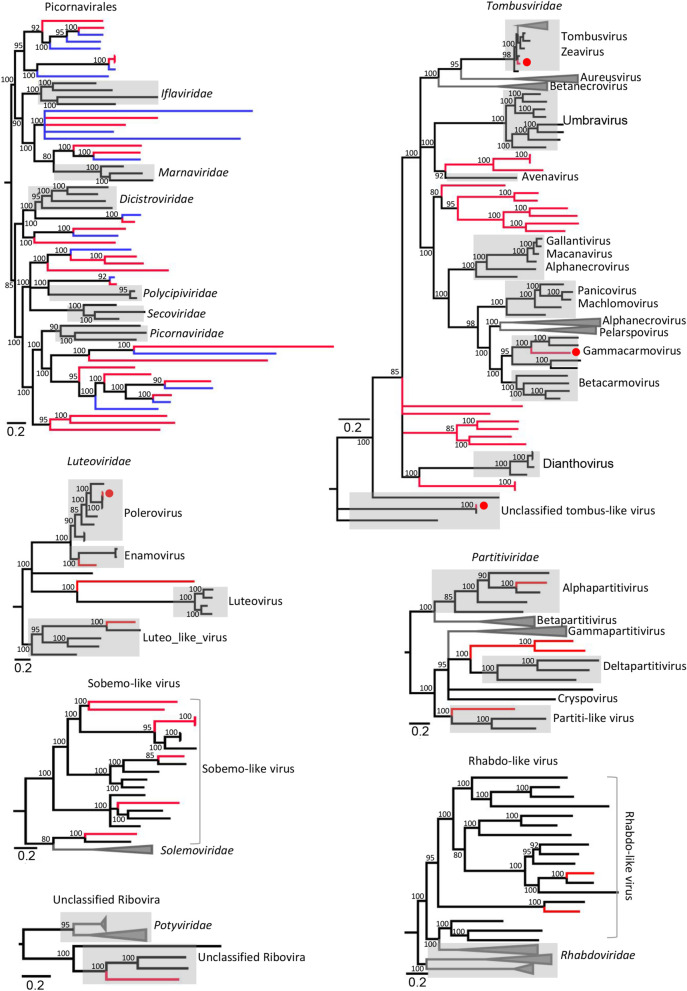
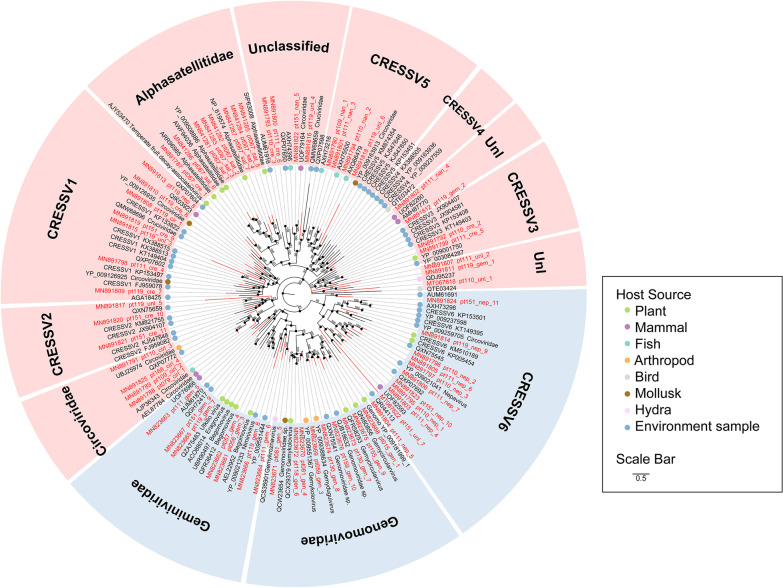
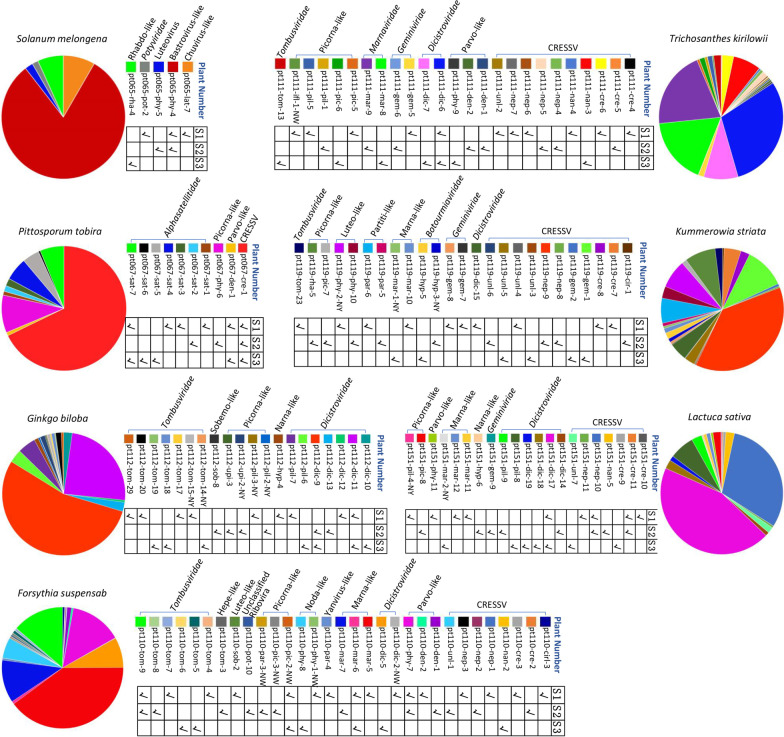
Results: We identified 245 distinct plant-associated virus genomes (88 DNA and 157 RNA viruses) belonging to 27 known viral families, orders, or unclassified virus groups. Some viral genomes were sufficiently divergent to comprise new species, genera, families, or even orders. Some groups of viruses were detected that currently are only known to infect organisms other than plants. It indicates a wider host range for members of these clades than previously recognized theoretically. We cannot rule out that some viruses could be from plant contaminating organisms, although some methods were taken to get rid of them as much as possible. The same viral species could be found in different plants and co-infections were common.
Conclusions: Our data describe a complex viral community within a single plant ecosystem and expand our understanding of plant-associated viral diversity and their possible host ranges.
Keywords: Co-infection; Phylogenetic analysis; Phytocommunity; Plant virome; Virus host switching.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources