

MyD88-dependent signaling drives toll-like receptor-induced trained immunity in macrophages
- PMID: 36439136
- PMCID: PMC9692127
- DOI: 10.3389/fimmu.2022.1044662
MyD88-dependent signaling drives toll-like receptor-induced trained immunity in macrophages
Abstract
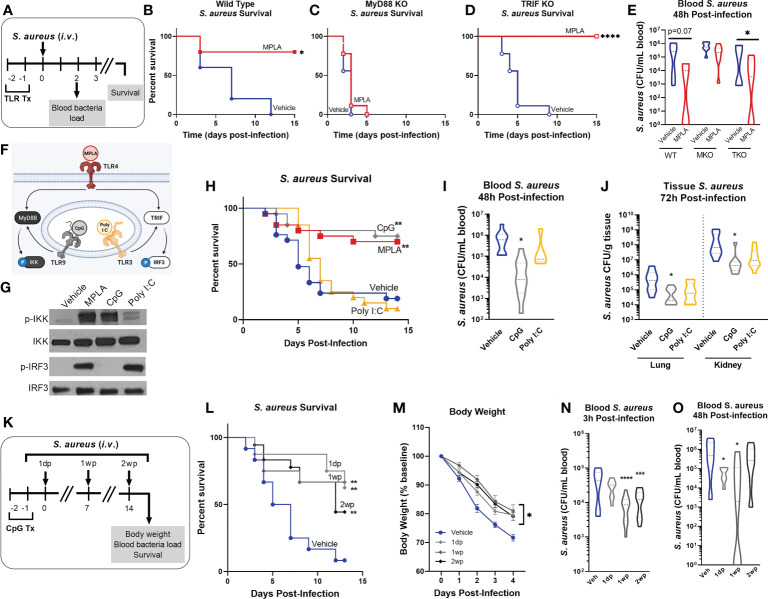
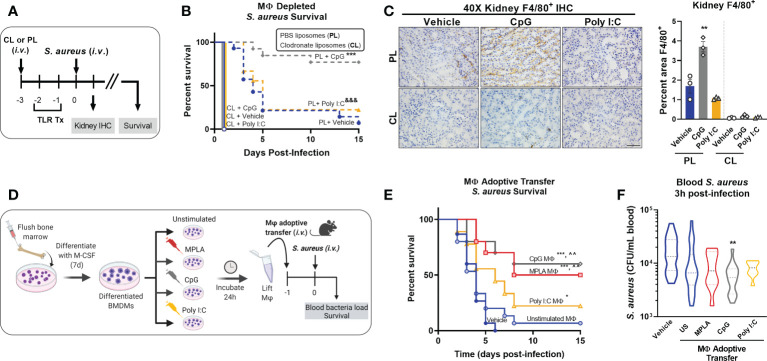
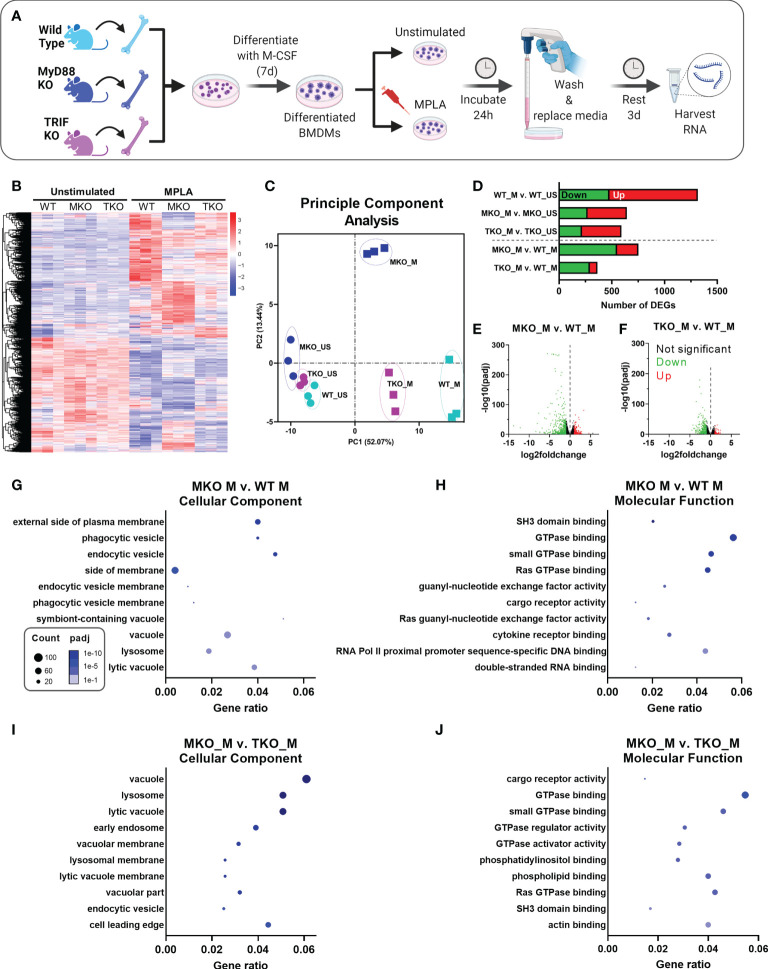
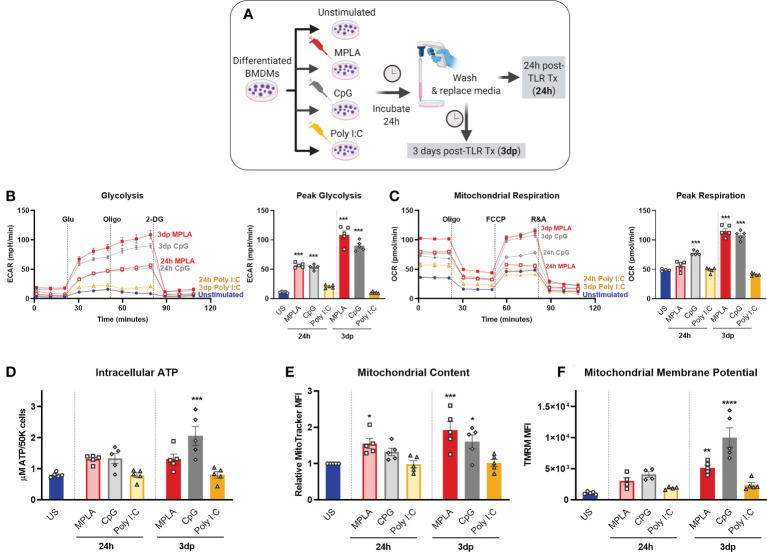
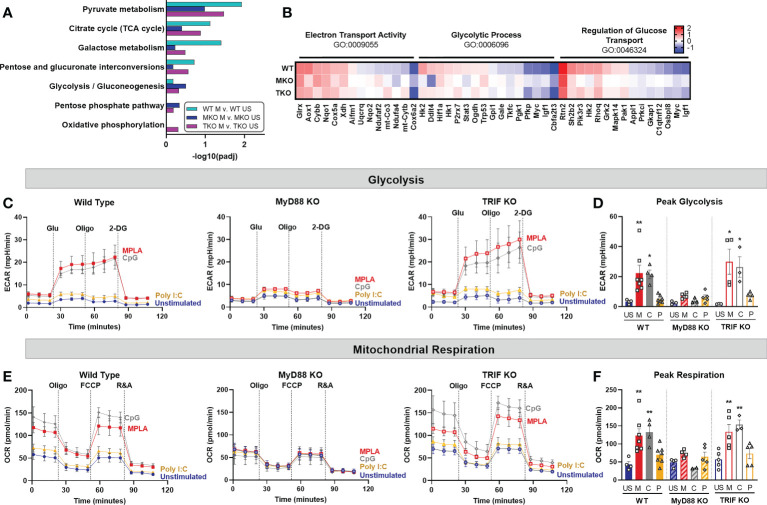
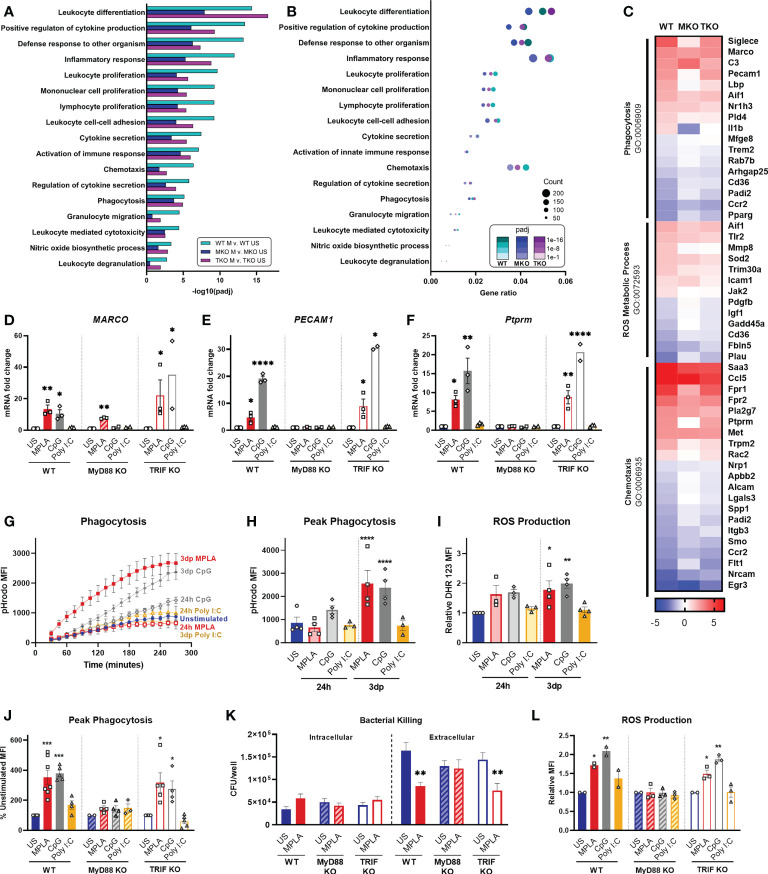
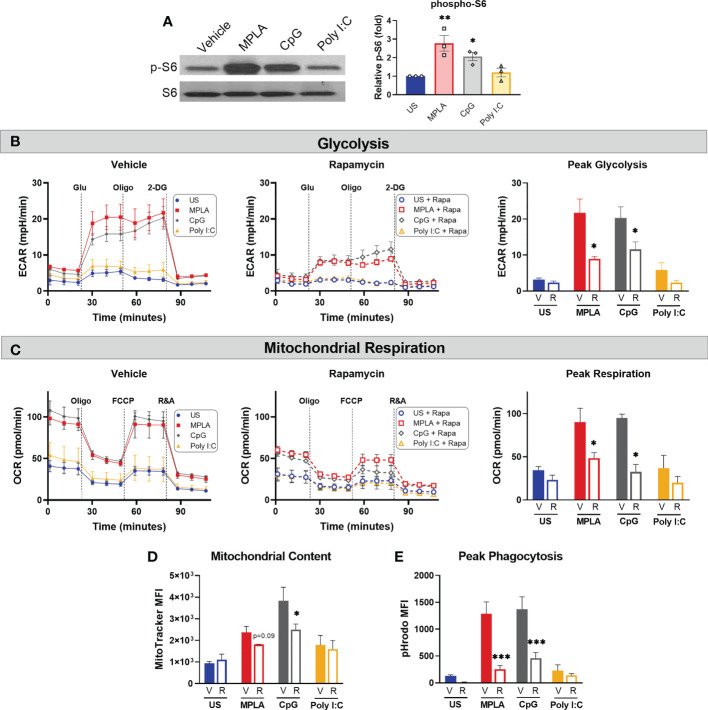
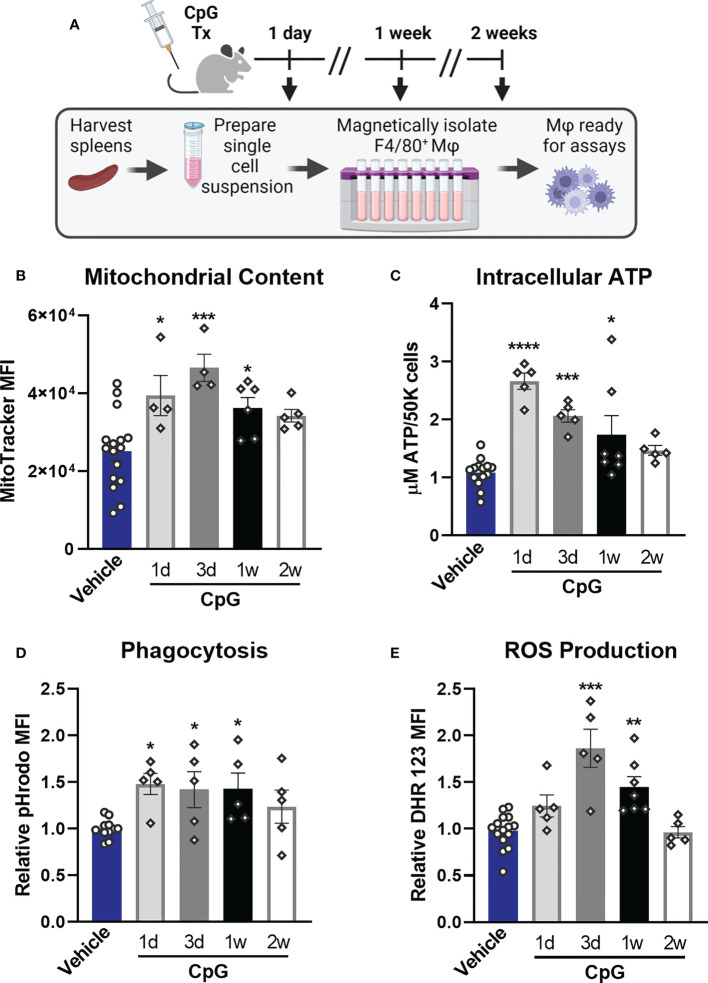
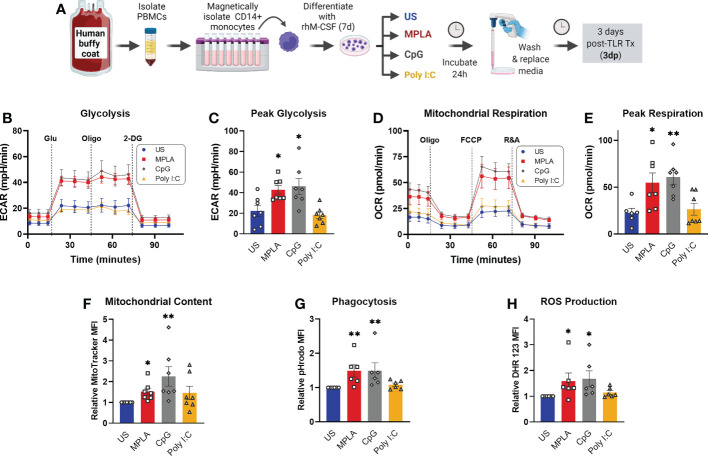
Immunocompromised populations are highly vulnerable to developing life-threatening infections. Strategies to protect patients with weak immune responses are urgently needed. Employing trained immunity, whereby innate leukocytes undergo reprogramming upon exposure to a microbial product and respond more robustly to subsequent infection, is a promising approach. Previously, we demonstrated that the TLR4 agonist monophosphoryl lipid A (MPLA) induces trained immunity and confers broad resistance to infection. TLR4 signals through both MyD88- and TRIF-dependent cascades, but the relative contribution of each pathway to induction of trained immunity is unknown. Here, we show that MPLA-induced resistance to Staphylococcus aureus infection is lost in MyD88-KO, but not TRIF-KO, mice. The MyD88-activating agonist CpG (TLR9 agonist), but not TRIF-activating Poly I:C (TLR3 agonist), protects against infection in a macrophage-dependent manner. MPLA- and CpG-induced augmentation of macrophage metabolism and antimicrobial functions is blunted in MyD88-, but not TRIF-KO, macrophages. Augmentation of antimicrobial functions occurs in parallel to metabolic reprogramming and is dependent, in part, on mTOR activation. Splenic macrophages from CpG-treated mice confirmed that TLR/MyD88-induced reprogramming occurs in vivo. TLR/MyD88-triggered metabolic and functional reprogramming was reproduced in human monocyte-derived macrophages. These data show that MyD88-dependent signaling is critical in TLR-mediated trained immunity.
Keywords: MyD88; TLR4; innate immune memory; innate immunity; macrophage; metabolic reprogramming; toll-like receptor (TLR); trained immunity.
Copyright © 2022 Owen, Luan, Burelbach, McBride, Stothers, Boykin, Sivanesam, Schaedel, Patil, Wang, Hernandez, Patil, Sherwood and Bohannon.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- CDC . Antibiotic resistance threats in the united states, 2019. Atlanta, GA: U.S. Department of Health and Human Services; (2019).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous