

The role of adjuvants in overcoming antibacterial resistance due to enzymatic drug modification
- PMID: 36439977
- PMCID: PMC9667779
- DOI: 10.1039/d2md00263a
The role of adjuvants in overcoming antibacterial resistance due to enzymatic drug modification
Abstract
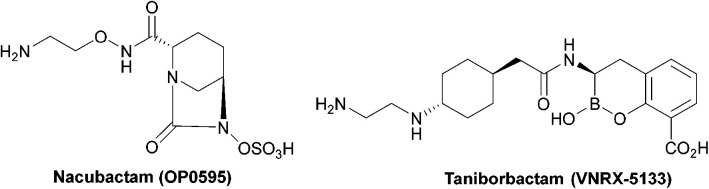
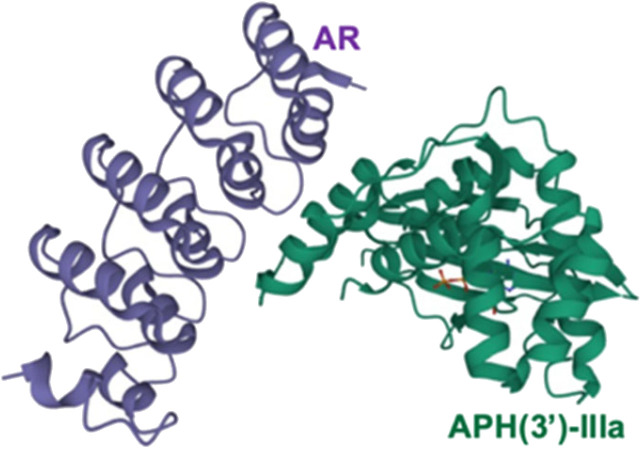
Antibacterial resistance is a prominent issue with monotherapy often leading to treatment failure in serious infections. Many mechanisms can lead to antibacterial resistance including deactivation of antibacterial agents by bacterial enzymes. Enzymatic drug modification confers resistance to β-lactams, aminoglycosides, chloramphenicol, macrolides, isoniazid, rifamycins, fosfomycin and lincosamides. Novel enzyme inhibitor adjuvants have been developed in an attempt to overcome resistance to these agents, only a few of which have so far reached the market. This review discusses the different enzymatic processes that lead to deactivation of antibacterial agents and provides an update on the current and potential enzyme inhibitors that may restore bacterial susceptibility.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
There are no conflicts of interest to declare.
Figures





























References
-
- W. H. O. (WHO), 2020 antibacterial agents in clinical and preclinical development: an overview and analysis, https://www.who.int/publications/i/item/9789240021303, (accessed May, 2022)
-
- W. H. O. (WHO), Antimicrobial resistance, https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistanc..., (accessed May, 2022)
Publication types
LinkOut - more resources
Full Text Sources