

ANKRD11 pathogenic variants and 16q24.3 microdeletions share an altered DNA methylation signature in patients with KBG syndrome
- PMID: 36440975
- PMCID: PMC10117159
- DOI: 10.1093/hmg/ddac289
ANKRD11 pathogenic variants and 16q24.3 microdeletions share an altered DNA methylation signature in patients with KBG syndrome
Abstract
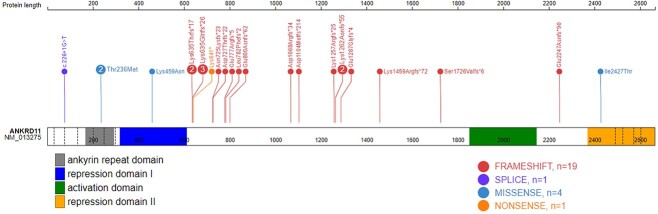
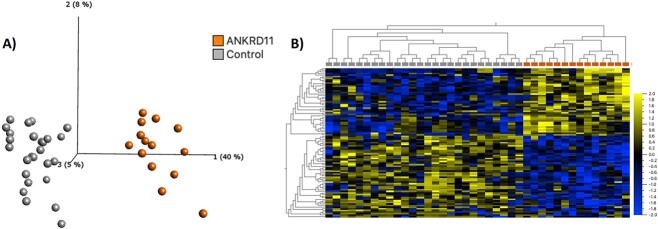
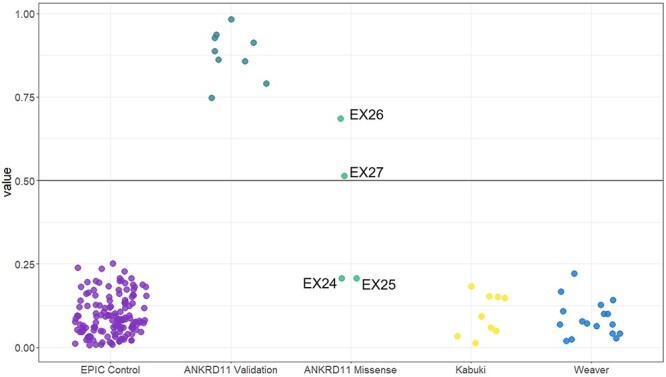
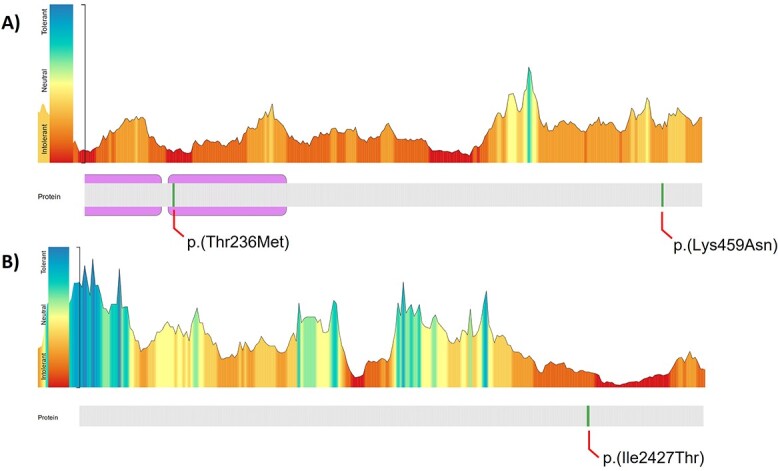
Pathogenic variants in ANKRD11 or microdeletions at 16q24.3 are the cause of KBG syndrome (KBGS), a neurodevelopmental syndrome characterized by intellectual disability, dental and skeletal anomalies, and characteristic facies. The ANKRD11 gene encodes the ankyrin repeat-containing protein 11A transcriptional regulator, which is expressed in the brain and implicated in neural development. Syndromic conditions caused by pathogenic variants in epigenetic regulatory genes show unique patterns of DNA methylation (DNAm) in peripheral blood, termed DNAm signatures. Given ANKRD11's role in chromatin modification, we tested whether pathogenic ANKRD11 variants underlying KBGS are associated with a DNAm signature. We profiled whole-blood DNAm in 21 individuals with ANKRD11 variants, 2 individuals with microdeletions at 16q24.3 and 28 typically developing individuals, using Illumina's Infinium EPIC array. We identified 95 differentially methylated CpG sites that distinguished individuals with KBGS and pathogenic variants in ANKRD11 (n = 14) from typically developing controls (n = 28). This DNAm signature was then validated in an independent cohort of seven individuals with KBGS and pathogenic ANKRD11 variants. We generated a machine learning model from the KBGS DNAm signature and classified the DNAm profiles of four individuals with variants of uncertain significance (VUS) in ANKRD11. We identified an intermediate classification score for an inherited missense variant transmitted from a clinically unaffected mother to her affected child. In conclusion, we show that the DNAm profiles of two individuals with 16q24.3 microdeletions were indistinguishable from the DNAm profiles of individuals with pathogenic variants in ANKRD11, and we demonstrate the diagnostic utility of the new KBGS signature by classifying the DNAm profiles of individuals with VUS in ANKRD11.
© The Author(s) 2022. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
Insights into the ANKRD11 variants and short-stature phenotype through literature review and ClinVar database search.Orphanet J Rare Dis. 2024 Aug 12;19(1):292. doi: 10.1186/s13023-024-03301-y. Orphanet J Rare Dis. 2024. PMID: 39135054 Free PMC article. Review.
-
Missense variants in ANKRD11 cause KBG syndrome by impairment of stability or transcriptional activity of the encoded protein.Genet Med. 2022 Oct;24(10):2051-2064. doi: 10.1016/j.gim.2022.06.007. Epub 2022 Jul 14. Genet Med. 2022. PMID: 35833929
-
Haploinsufficiency for ANKRD11-flanking genes makes the difference between KBG and 16q24.3 microdeletion syndromes: 12 new cases.Eur J Hum Genet. 2017 Jun;25(6):694-701. doi: 10.1038/ejhg.2017.49. Epub 2017 Apr 19. Eur J Hum Genet. 2017. PMID: 28422132 Free PMC article.
-
Clinical and molecular findings in 39 patients with KBG syndrome caused by deletion or mutation of ANKRD11.Am J Med Genet A. 2016 Nov;170(11):2847-2859. doi: 10.1002/ajmg.a.37878. Epub 2016 Sep 8. Am J Med Genet A. 2016. PMID: 27605097
-
KBG syndrome: Clinical features and molecular findings in seven unrelated Korean families with a review of the literature.Mol Genet Genomic Med. 2023 Apr;11(4):e2127. doi: 10.1002/mgg3.2127. Epub 2022 Dec 23. Mol Genet Genomic Med. 2023. PMID: 36564961 Free PMC article. Review.
Cited by
-
Episignatures in practice: independent evaluation of published episignatures for the molecular diagnostics of ten neurodevelopmental disorders.Eur J Hum Genet. 2024 Feb;32(2):190-199. doi: 10.1038/s41431-023-01474-x. Epub 2023 Oct 23. Eur J Hum Genet. 2024. PMID: 37872275 Free PMC article.
-
SRSF1 haploinsufficiency is responsible for a syndromic developmental disorder associated with intellectual disability.Am J Hum Genet. 2023 May 4;110(5):790-808. doi: 10.1016/j.ajhg.2023.03.016. Epub 2023 Apr 17. Am J Hum Genet. 2023. PMID: 37071997 Free PMC article.
-
Pathogenic variants in KMT2C result in a neurodevelopmental disorder distinct from Kleefstra and Kabuki syndromes.Am J Hum Genet. 2024 Aug 8;111(8):1626-1642. doi: 10.1016/j.ajhg.2024.06.009. Epub 2024 Jul 15. Am J Hum Genet. 2024. PMID: 39013459 Free PMC article.
-
Functional investigation of a novel ANKRD11 frameshift variant identified in a Chinese family with KBG syndrome.Heliyon. 2024 Mar 13;10(6):e28082. doi: 10.1016/j.heliyon.2024.e28082. eCollection 2024 Mar 30. Heliyon. 2024. PMID: 38515699 Free PMC article.
-
Insights into the ANKRD11 variants and short-stature phenotype through literature review and ClinVar database search.Orphanet J Rare Dis. 2024 Aug 12;19(1):292. doi: 10.1186/s13023-024-03301-y. Orphanet J Rare Dis. 2024. PMID: 39135054 Free PMC article. Review.
References
-
- Zhang, A., Yeung, P.L., Li, C.W., Tsai, S.C., Dinh, G.K., Wu, X., Li, H. and Chen, J.D. (2004) Identification of a novel family of ankyrin repeats containing cofactors for p160 nuclear receptor coactivators. J. Biol. Chem., 279, 33799–33805. - PubMed
-
- Herrmann, J., Pallister, P.D., Tiddy, W. and Opitz, J.M. (1975) The KBG syndrome-a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects Orig. Artic. Ser., 11, 7–18. - PubMed
-
- Sirmaci, A., Spiliopoulos, M., Brancati, F., Powell, E., Duman, D., Abrams, A., Bademci, G., Agolini, E., Guo, S., Konuk, B.et al. (2011) Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am. J. Hum. Genet., 89, 289–294. - PMC - PubMed
