

Transcriptome-based molecular subtypes and differentiation hierarchies improve the classification framework of acute myeloid leukemia
- PMID: 36442087
- PMCID: PMC9894241
- DOI: 10.1073/pnas.2211429119
Transcriptome-based molecular subtypes and differentiation hierarchies improve the classification framework of acute myeloid leukemia
Abstract
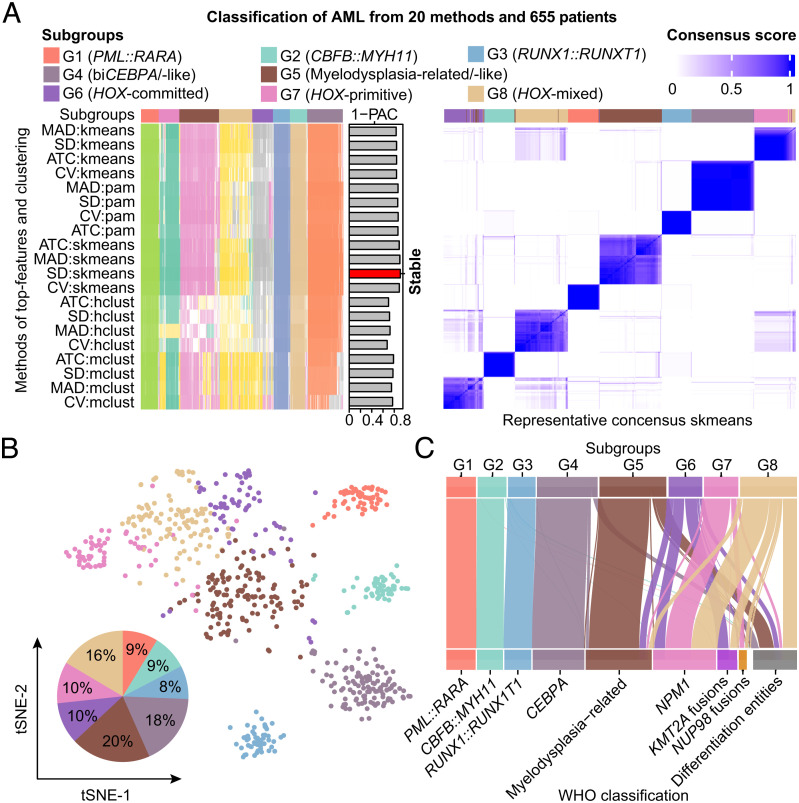
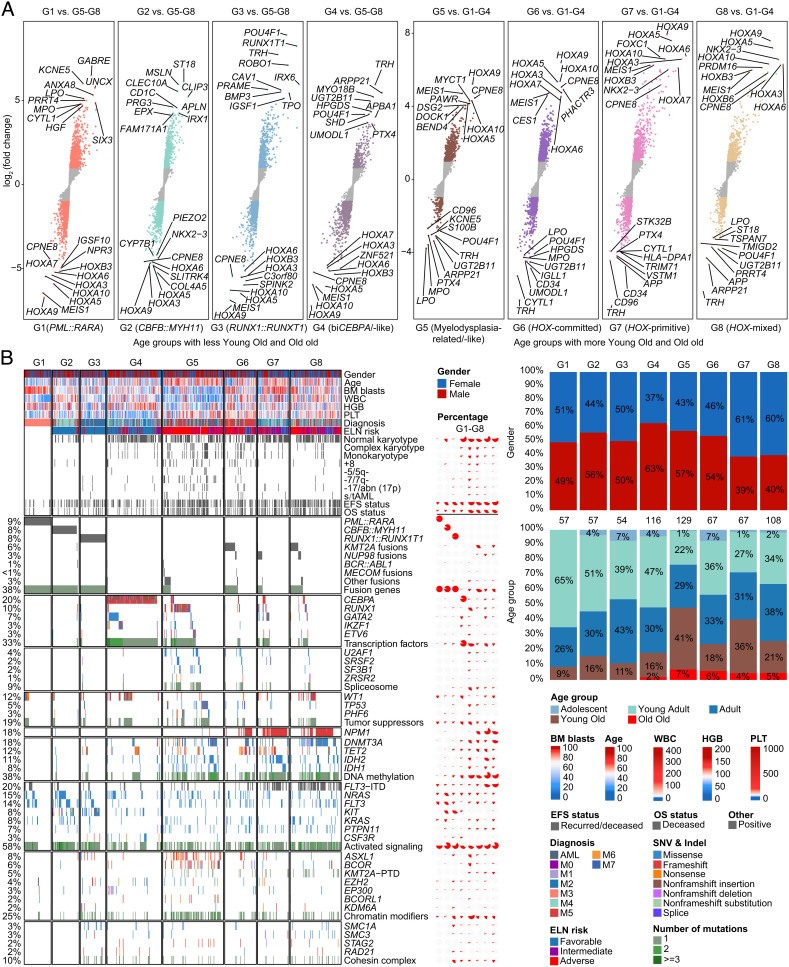
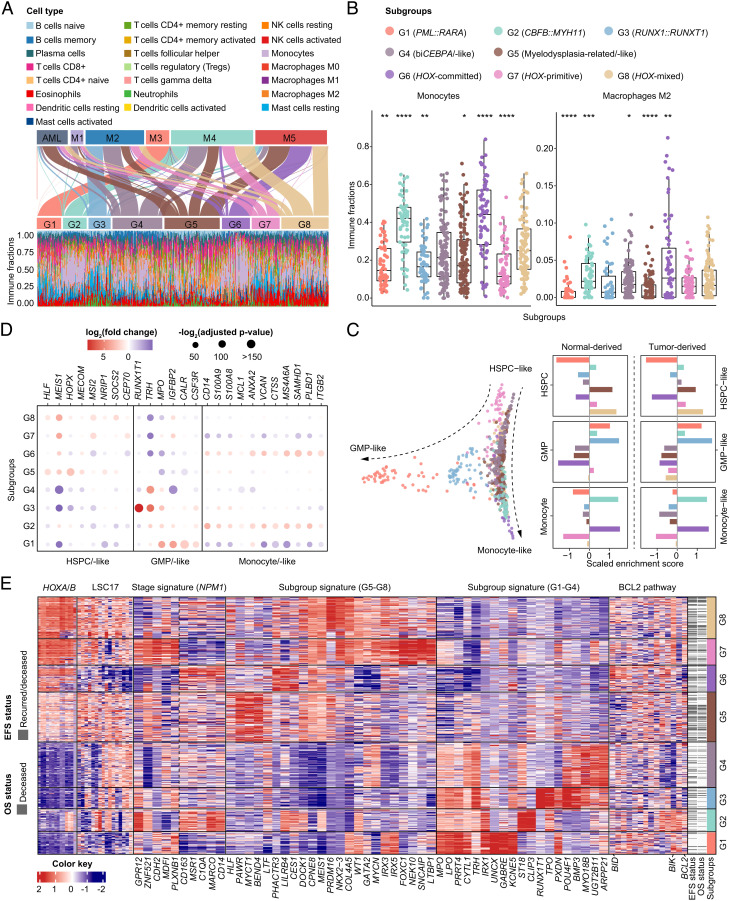
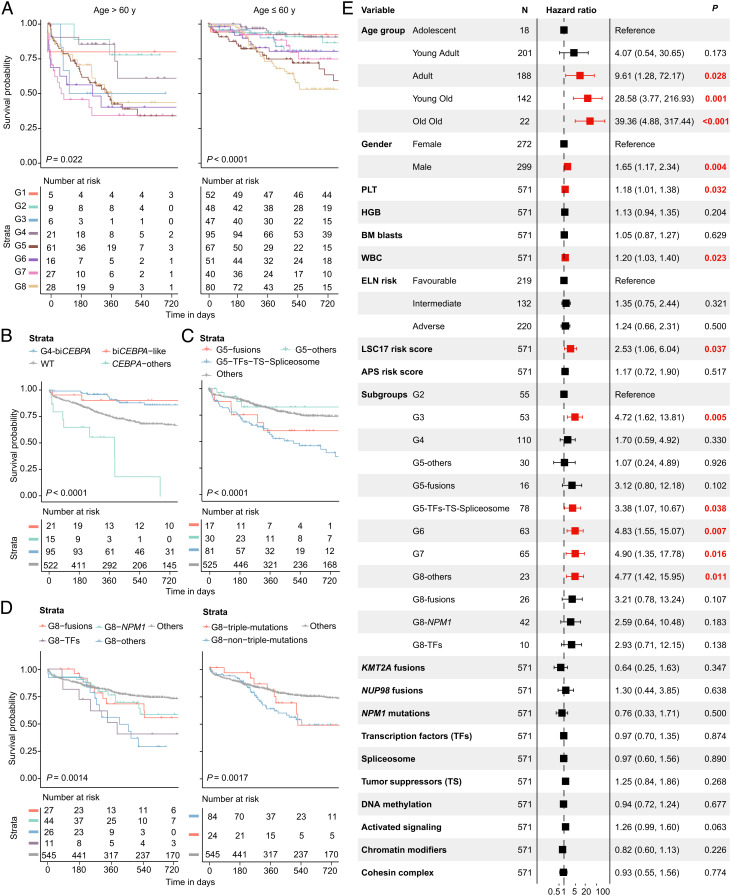
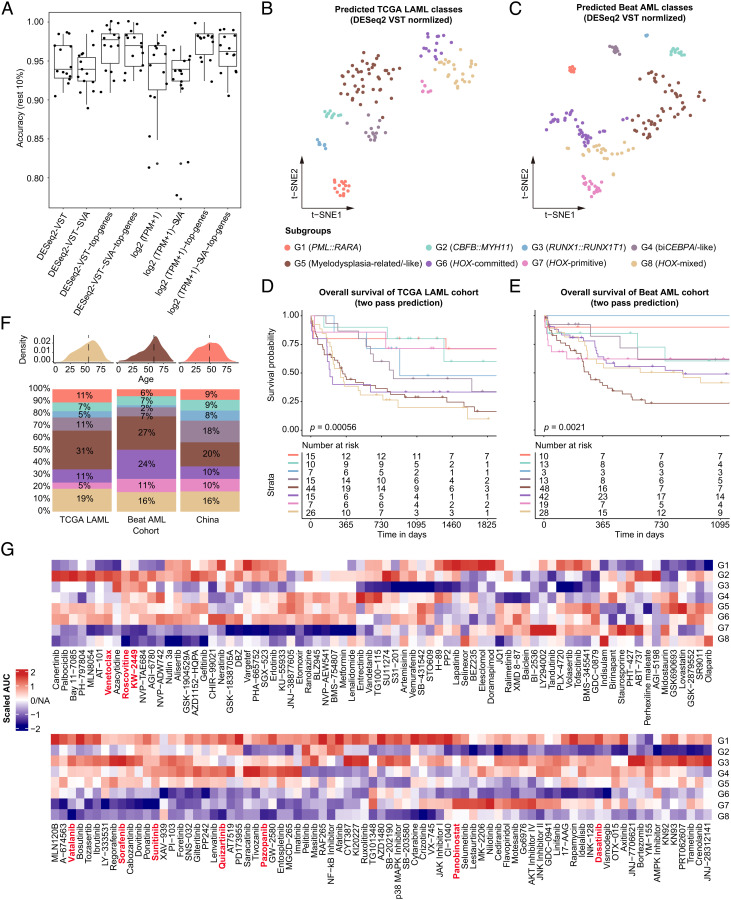
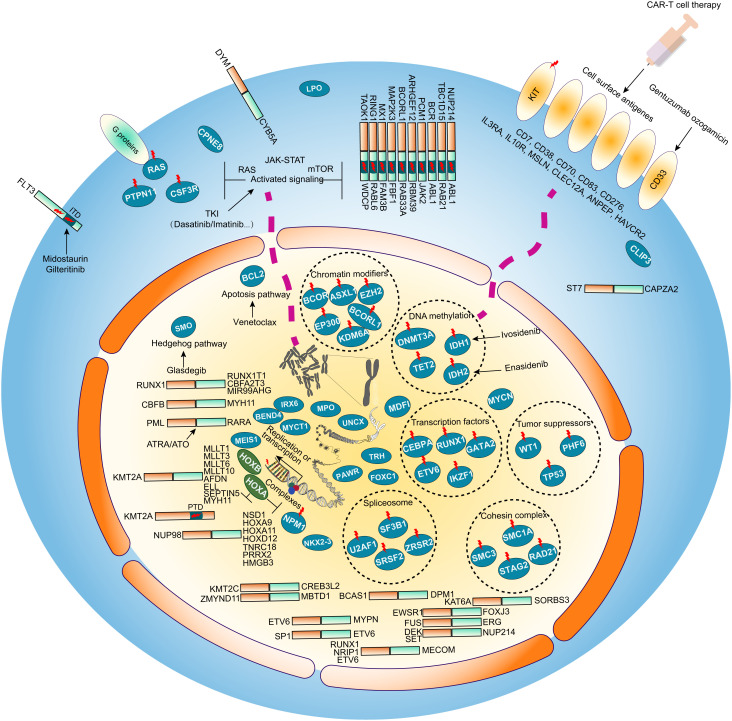
The current classification of acute myeloid leukemia (AML) relies largely on genomic alterations. Robust identification of clinically and biologically relevant molecular subtypes from nongenomic high-throughput sequencing data remains challenging. We established the largest multicenter AML cohort (n = 655) in China, with all patients subjected to RNA sequencing (RNA-Seq) and 619 (94.5%) to targeted or whole-exome sequencing (TES/WES). Based on an enhanced consensus clustering, eight stable gene expression subgroups (G1-G8) with unique clinical and biological significance were identified, including two unreported (G5 and G8) and three redefined ones (G4, G6, and G7). Apart from four well-known low-risk subgroups including PML::RARA (G1), CBFB::MYH11 (G2), RUNX1::RUNX1T1 (G3), biallelic CEBPA mutations or -like (G4), four meta-subgroups with poor outcomes were recognized. The G5 (myelodysplasia-related/-like) subgroup enriched clinical, cytogenetic and genetic features mimicking secondary AML, and hotspot mutations of IKZF1 (p.N159S) (n = 7). In contrast, most NPM1 mutations and KMT2A and NUP98 fusions clustered into G6-G8, showing high expression of HOXA/B genes and diverse differentiation stages, from hematopoietic stem/progenitor cell down to monocyte, namely HOX-primitive (G7), HOX-mixed (G8), and HOX-committed (G6). Through constructing prediction models, the eight gene expression subgroups could be reproduced in the Cancer Genome Atlas (TCGA) and Beat AML cohorts. Each subgroup was associated with distinct prognosis and drug sensitivities, supporting the clinical applicability of this transcriptome-based classification of AML. These molecular subgroups illuminate the complex molecular network of AML, which may promote systematic studies of disease pathogenesis and foster the screening of targeted agents based on omics.
Keywords: RNA-Seq; acute myeloid leukemia; cell differentiation; drug sensitivity; molecular classification.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Newell L. F., Cook R. J., Advances in acute myeloid leukemia. BMJ 375, n2026 (2021). - PubMed
-
- Charrot S., Armes H., Rio-Machin A., Fitzgibbon J., AML through the prism of molecular genetics. Br. J. Haematol. 188, 49–62 (2020). - PubMed
-
- Perl A. E., et al. , Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N. Engl. J. Med. 381, 1728–1740 (2019). - PubMed
-
- DiNardo C. D., et al. , Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N. Engl. J. Med. 383, 617–629 (2020). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
