

CCAR2 functions downstream of the Shieldin complex to promote double-strand break end-joining
- PMID: 36442094
- PMCID: PMC9894118
- DOI: 10.1073/pnas.2214935119
CCAR2 functions downstream of the Shieldin complex to promote double-strand break end-joining
Abstract
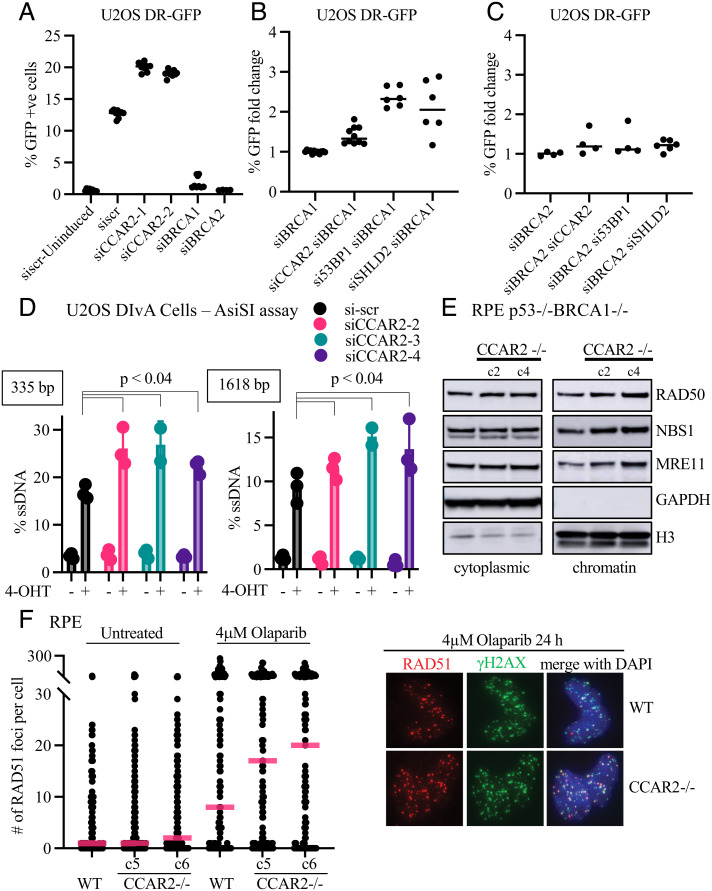
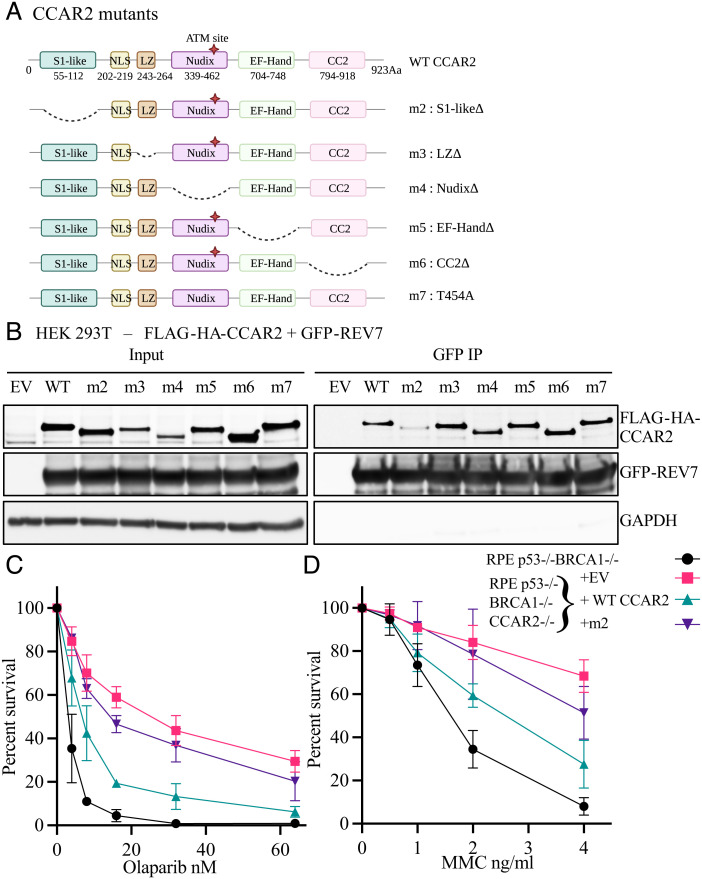
The 53BP1-RIF1 pathway restricts the resection of DNA double-strand breaks (DSBs) and promotes blunt end-ligation by non-homologous end joining (NHEJ) repair. The Shieldin complex is a downstream effector of the 53BP1-RIF1 pathway. Here, we identify a component of this pathway, CCAR2/DBC1, which is also required for restriction of DNA end-resection. CCAR2 co-immunoprecipitates with the Shieldin complex, and knockout of CCAR2 in a BRCA1-deficient cell line results in elevated DSB end-resection, RAD51 loading, and PARP inhibitor (PARPi) resistance. Knockout of CCAR2 is epistatic with knockout of other Shieldin proteins. The S1-like RNA-binding domain of CCAR2 is required for its interaction with the Shieldin complex and for suppression of DSB end-resection. CCAR2 functions downstream of the Shieldin complex, and CCAR2 knockout cells have delayed resolution of Shieldin complex foci. Forkhead-associated (FHA)-dependent targeting of CCAR2 to DSB sites re-sensitized BRCA1-/-SHLD2-/- cells to PARPi. Taken together, CCAR2 is a functional component of the 53BP1-RIF1 pathway, promotes the refill of resected DSBs, and suppresses homologous recombination.
Keywords: 53BP1; CCAR2/DBC1; Shieldin complex; homologous recombination; single nucleotide variants.
Conflict of interest statement
The authors declare no competing interest. A.D.D. reports consulting for Acerand Therapeutics, AstraZeneca, Bayer AG, Blacksmith/Lightstone Ventures, Bristol Myers Squibb, Cedilla Therapeutics, Constellation Pharmaceuticals, Cyteir Therapeutics, EMD Serono, Epizyme, Faze Medicines, GlaxoSmithKline, Impact Therapeutics, LAV Global Management Company Limited, PrimeFour Therapeutics, Patheon Pharmaceuticals, Pfizer, Tango Therapeutics, and Zentalis Pharmaceuticals/Zeno Management; is an Advisory Board member for Cedilla Therapeutics, Cyteir, and Impact Therapeutics; stockholder in Cedilla Therapeutics, Cyteir, Impact Therapeutics, and PrimeFour Therapeutics; and reports receiving commercial research grants from Bristol Myers Squibb, EMD Serono, Moderna, and Tango Therapeutics.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
