

Tumor-targeted delivery of a STING agonist improvescancer immunotherapy
- PMID: 36442099
- PMCID: PMC9894229
- DOI: 10.1073/pnas.2214278119
Tumor-targeted delivery of a STING agonist improvescancer immunotherapy
Abstract
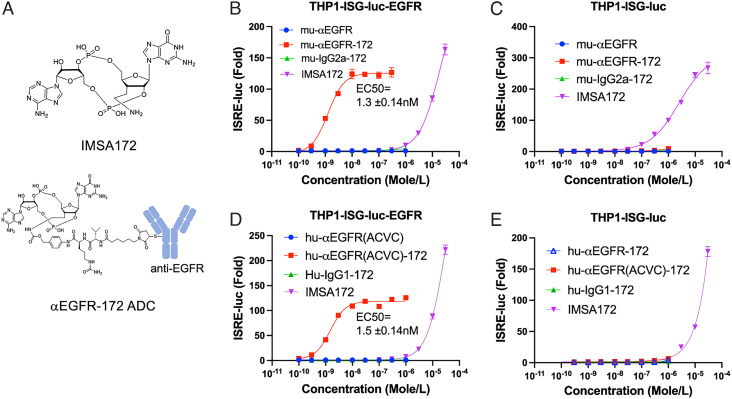
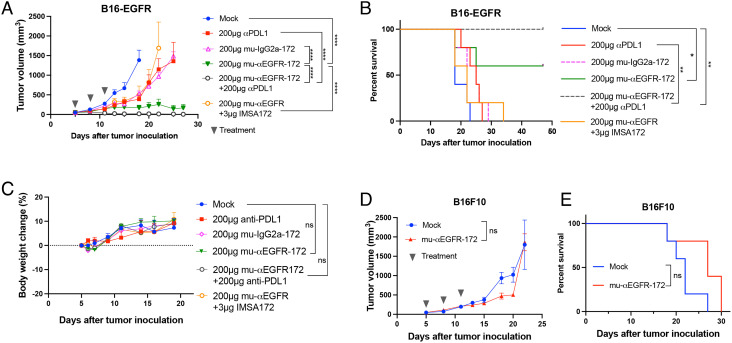
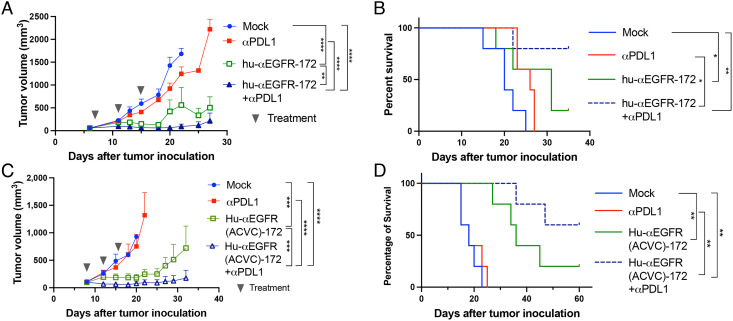
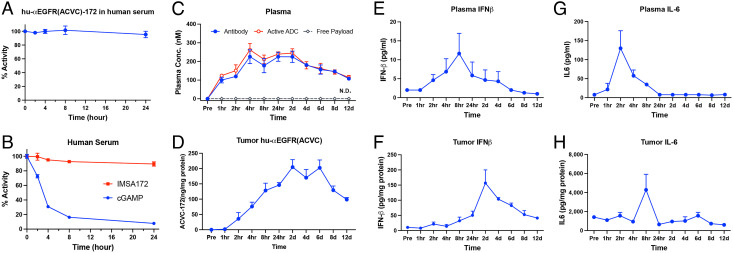
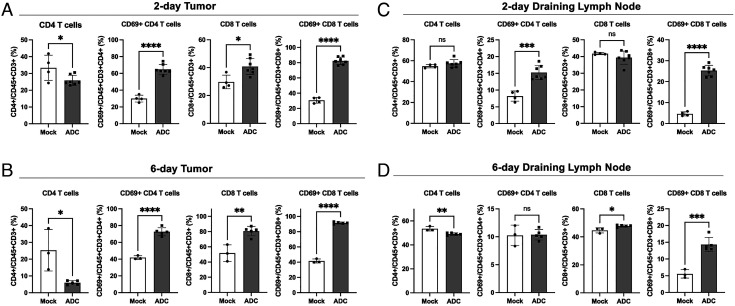
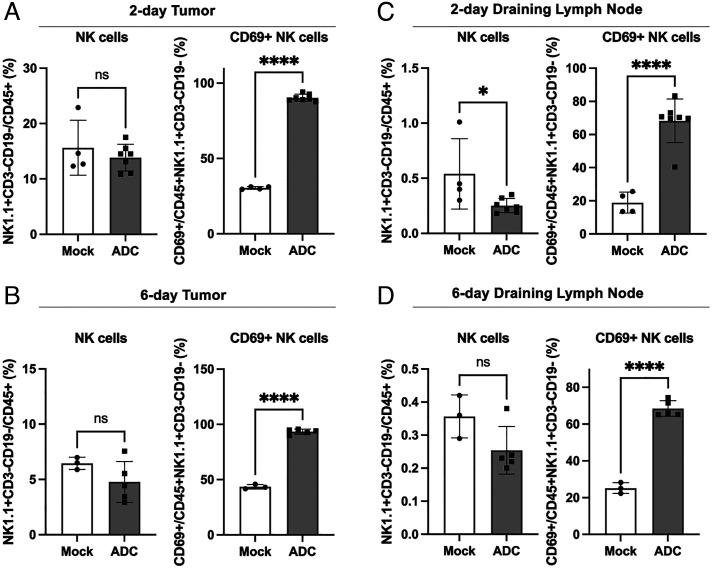
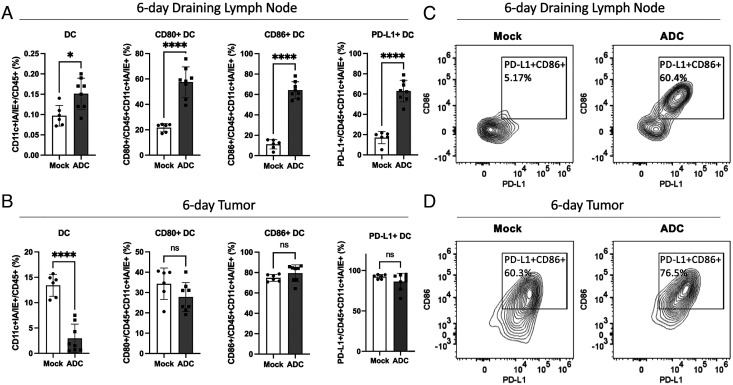
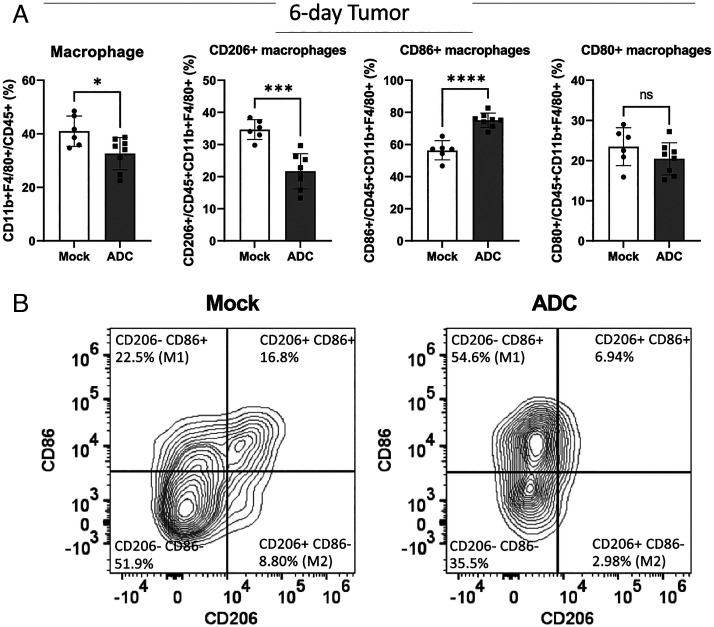
The cGAS-STING pathway is essential for immune defense against microbial pathogens and malignant cells; as such, STING is an attractive target for cancer immunotherapy. However, systemic administration of STING agonists poses safety issues while intratumoral injection is limited by tumor accessibility. Here, we generated antibody-drug conjugates (ADCs) by conjugating a STING agonist through a cleavable linker to antibodies targeting tumor cells. Systemic administration of these ADCs was well tolerated and exhibited potent antitumor efficacy in syngeneic mouse tumor models. The STING ADC further synergized with an anti-PD-L1 antibody to achieve superior antitumor efficacy. The STING ADC promoted multiple aspects of innate and adaptive antitumor immune responses, including activation of dendritic cells, T cells, natural killer cells and natural killer T cells, as well as promotion of M2 to M1 polarization of tumor-associated macrophages. These results provided the proof of concept for clinical development of the STING ADCs.
Keywords: ADC; STING; cGAS; cancer; tumor immunity.
Conflict of interest statement
The authors declare a competing interest. The authors have organizational affiliations to disclose, Z.J.C. is a scientific collaborator with Pfizer and ImmuneSensor Therapeutics, and a scientific advisor for Brii Biosciences, Genor Biopharma and Drug Farm. C.C. is a scientific advisor for ImmuneSensor Therapeutics., Z.J.C. has stock ownership in Brii Biosciences. H.S., L.S., C.C., and Z.J.C. are inventors of US patent 10,336,786. Q.W., H.S., L.S, C.C., and Z.J.C. are inventors of US patent 10,519,188. Y.-t.W., Q.W., H.S., J.Q, L.S., C.C., and Z.J.C. are inventors of US patent 11,033,635. Z.J.C. received research support from Pfizer and ImmuneSensor Therapeutics.
Figures
References
-
- O’Donnell J. S., Teng M. W. L., Smyth M. J., Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 16, 151–167 (2019). - PubMed
-
- Bagchi S., Yuan R., Engleman E. G., Immune checkpoint inhibitors for the treatment of cancer: Clinical impact and mechanisms of response and resistance. Annu. Rev. Pathol. 16, 223–249 (2021). - PubMed
-
- Ablasser A., Chen Z. J., cGAS in action: Expanding roles in immunity and inflammation. Science 363, eaat8657 (2019). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
