

Experimental evidence for temporal uncoupling of brain Aβ deposition and neurodegenerative sequelae
- PMID: 36443293
- PMCID: PMC9705543
- DOI: 10.1038/s41467-022-34538-5
Experimental evidence for temporal uncoupling of brain Aβ deposition and neurodegenerative sequelae
Abstract
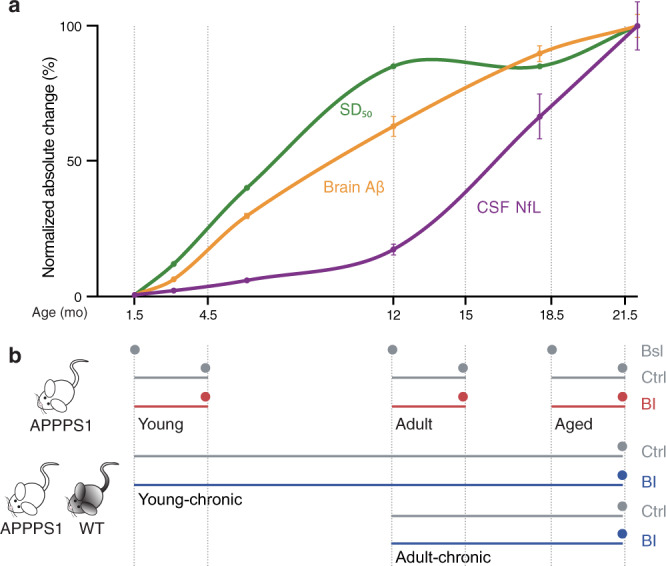
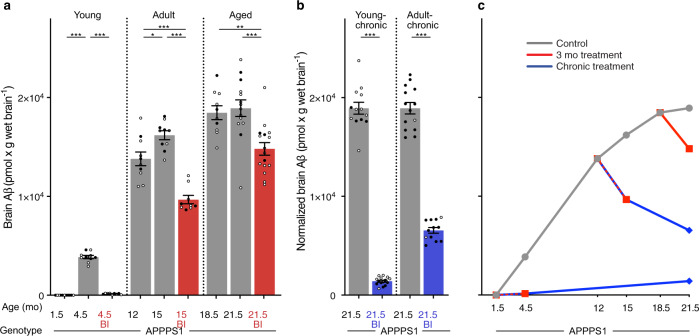
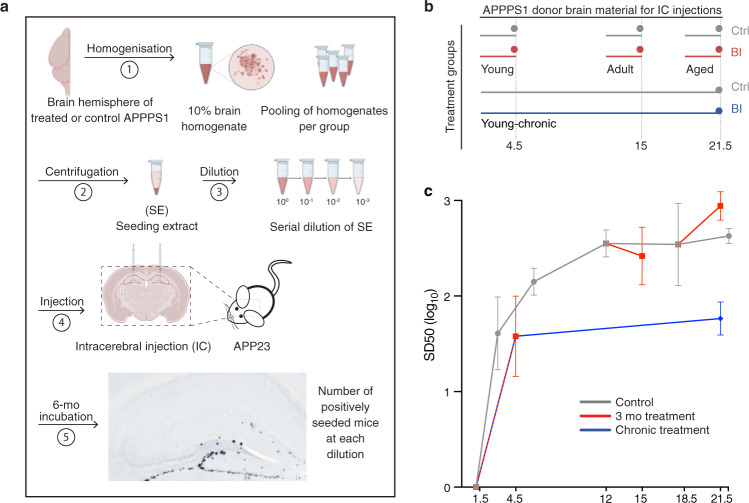
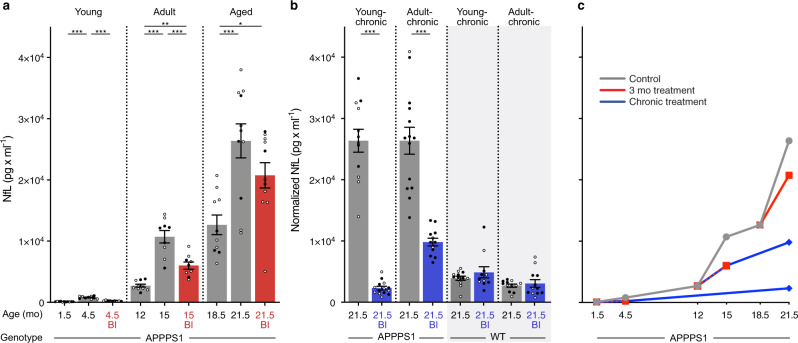
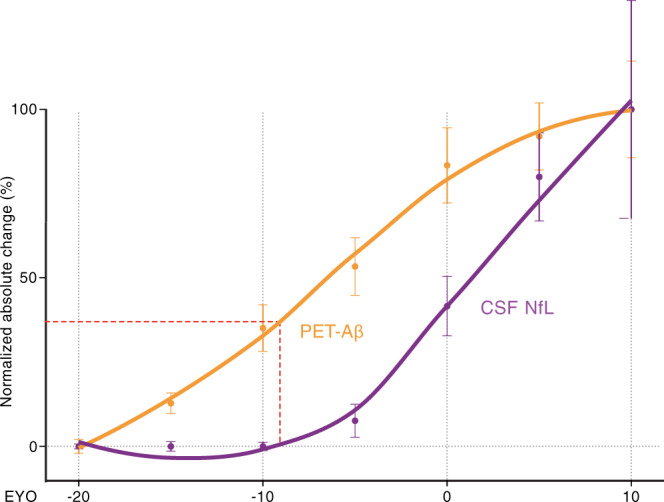
Brain Aβ deposition is a key early event in the pathogenesis of Alzheimer´s disease (AD), but the long presymptomatic phase and poor correlation between Aβ deposition and clinical symptoms remain puzzling. To elucidate the dependency of downstream pathologies on Aβ, we analyzed the trajectories of cerebral Aβ accumulation, Aβ seeding activity, and neurofilament light chain (NfL) in the CSF (a biomarker of neurodegeneration) in Aβ-precursor protein transgenic mice. We find that Aβ deposition increases linearly until it reaches an apparent plateau at a late age, while Aβ seeding activity increases more rapidly and reaches a plateau earlier, coinciding with the onset of a robust increase of CSF NfL. Short-term inhibition of Aβ generation in amyloid-laden mice reduced Aβ deposition and associated glial changes, but failed to reduce Aβ seeding activity, and CSF NfL continued to increase although at a slower pace. When short-term or long-term inhibition of Aβ generation was started at pre-amyloid stages, CSF NfL did not increase despite some Aβ deposition, microglial activation, and robust brain Aβ seeding activity. A dissociation of Aβ load and CSF NfL trajectories was also found in familial AD, consistent with the view that Aβ aggregation is not kinetically coupled to neurotoxicity. Rather, neurodegeneration starts when Aβ seeding activity is saturated and before Aβ deposition reaches critical (half-maximal) levels, a phenomenon reminiscent of the two pathogenic phases in prion disease.
© 2022. The Author(s).
Conflict of interest statement
I.B., D.S., U.N. were employees of Novartis at the time the study was conducted. M.J. has received consulting fees from Papiling GmbH. All the other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
