

Standardized multi-omics of Earth's microbiomes reveals microbial and metabolite diversity
- PMID: 36443458
- PMCID: PMC9712116
- DOI: 10.1038/s41564-022-01266-x
Standardized multi-omics of Earth's microbiomes reveals microbial and metabolite diversity
Abstract
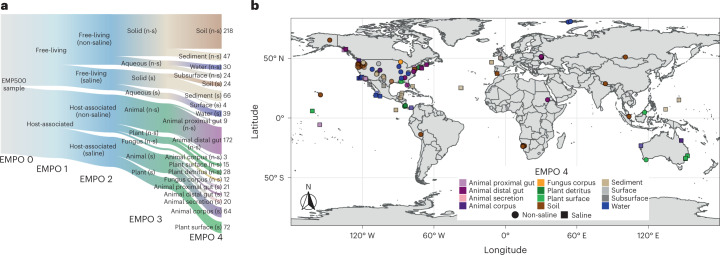
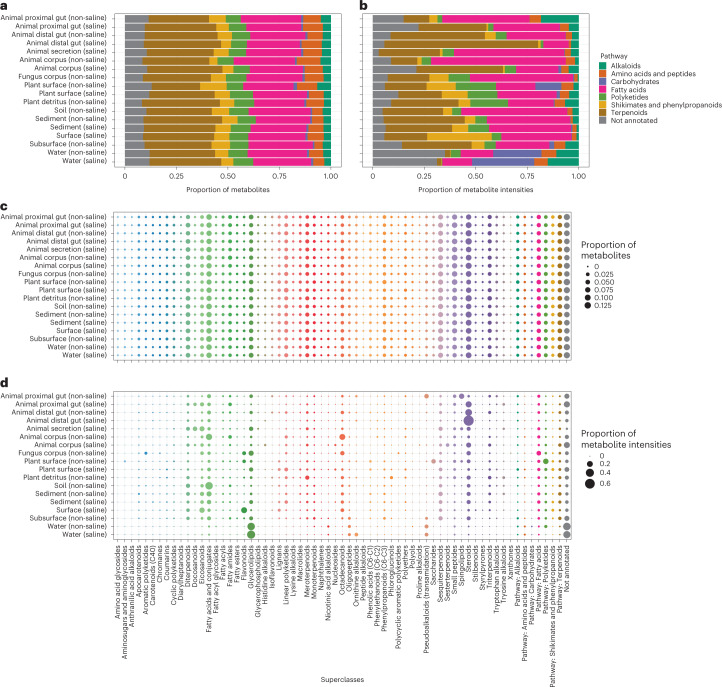
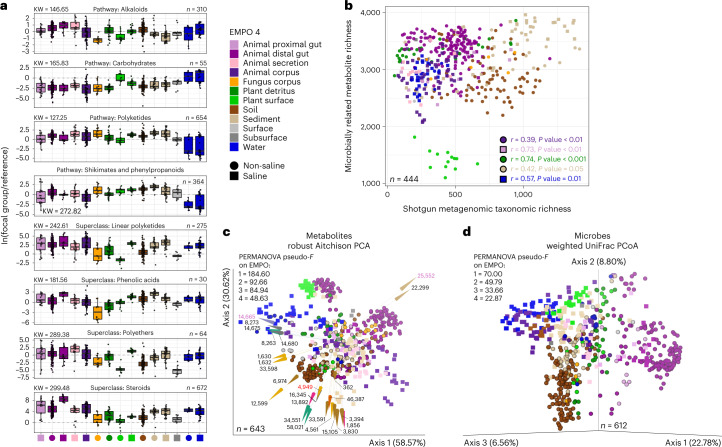
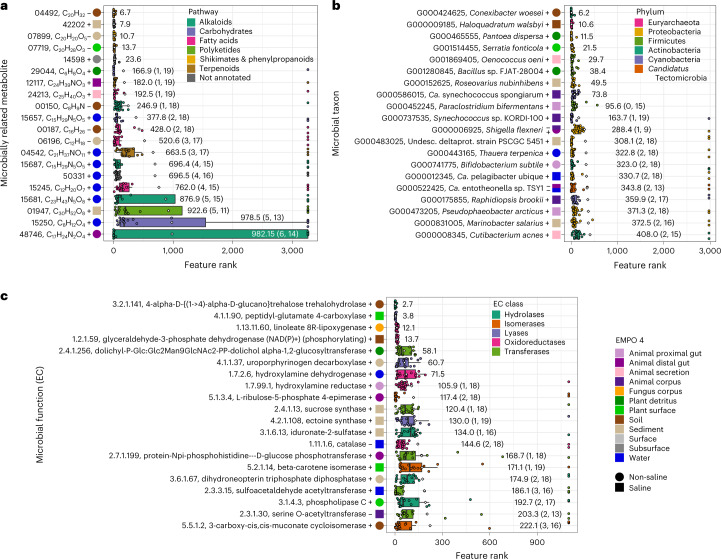
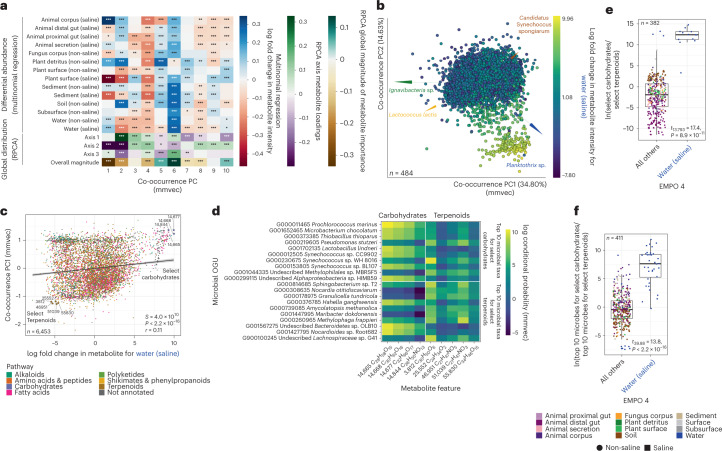
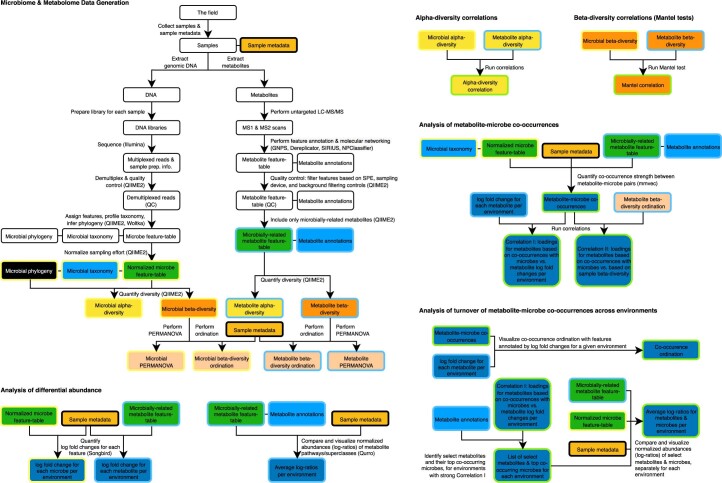
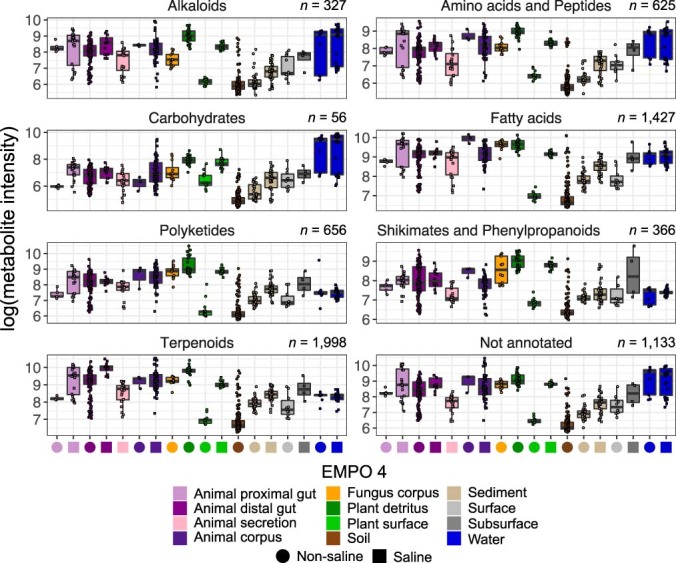
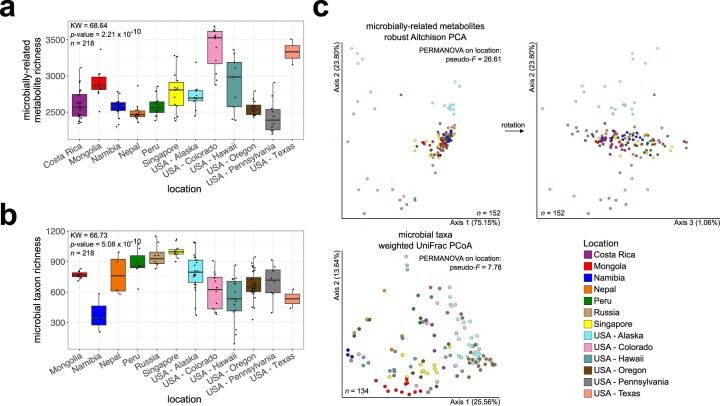
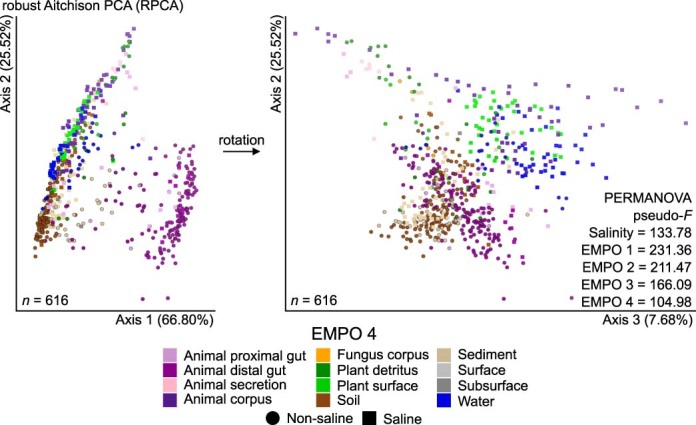
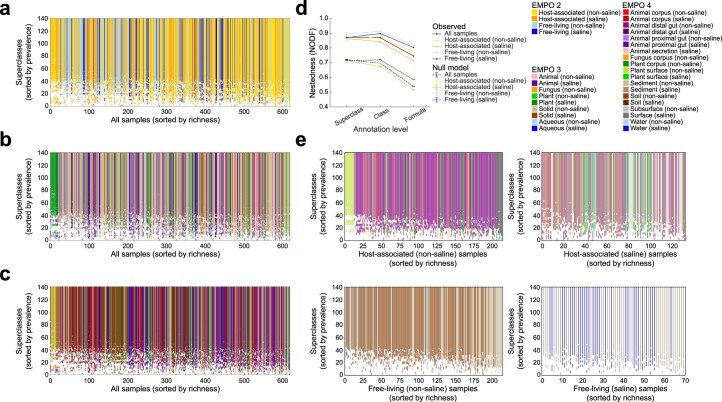
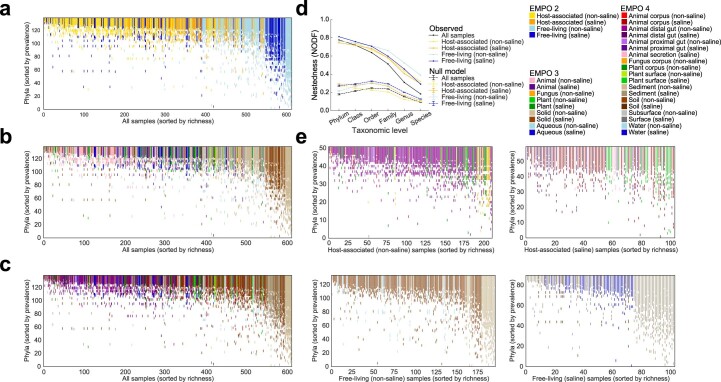
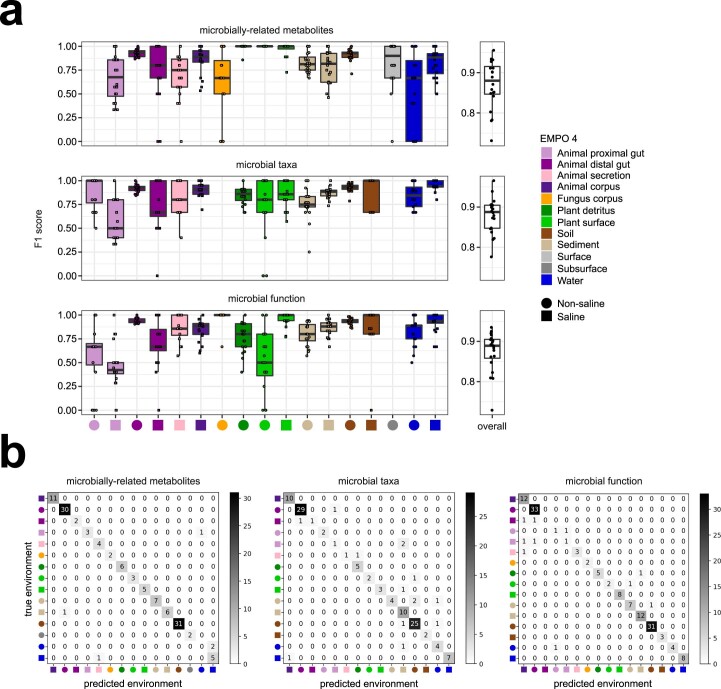
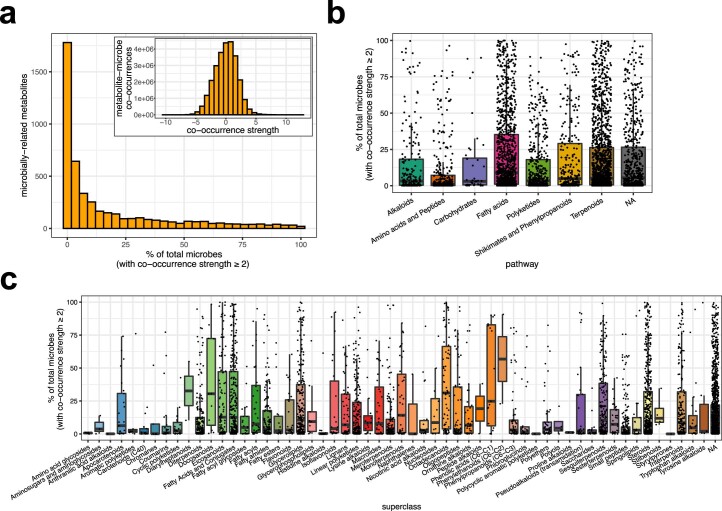
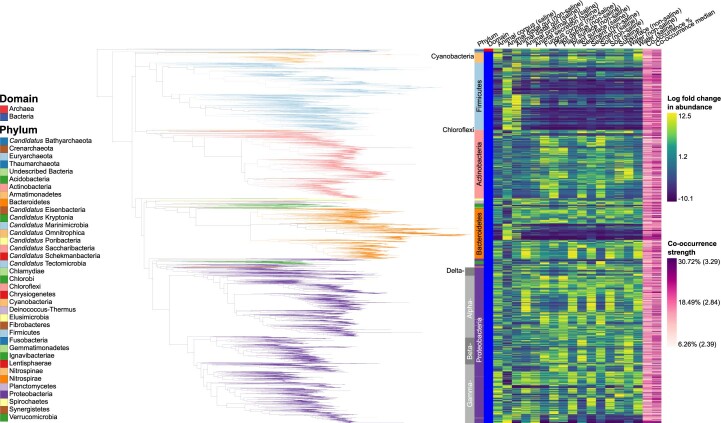
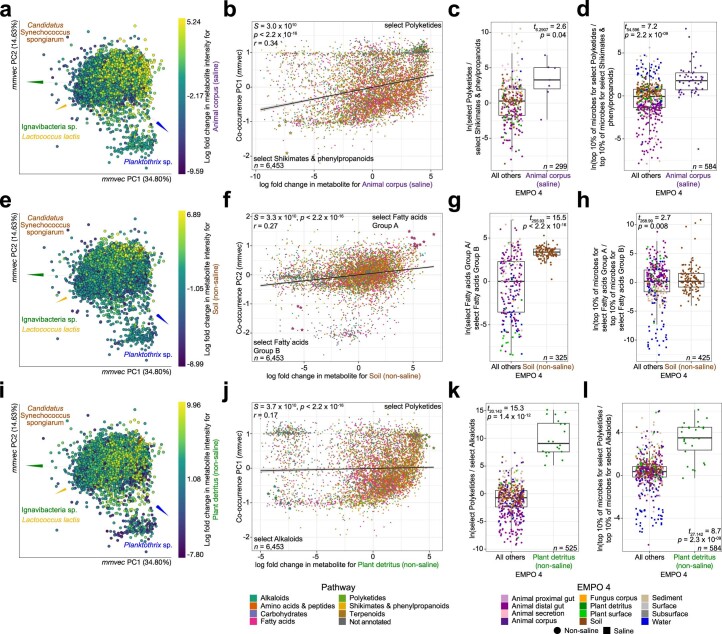
Despite advances in sequencing, lack of standardization makes comparisons across studies challenging and hampers insights into the structure and function of microbial communities across multiple habitats on a planetary scale. Here we present a multi-omics analysis of a diverse set of 880 microbial community samples collected for the Earth Microbiome Project. We include amplicon (16S, 18S, ITS) and shotgun metagenomic sequence data, and untargeted metabolomics data (liquid chromatography-tandem mass spectrometry and gas chromatography mass spectrometry). We used standardized protocols and analytical methods to characterize microbial communities, focusing on relationships and co-occurrences of microbially related metabolites and microbial taxa across environments, thus allowing us to explore diversity at extraordinary scale. In addition to a reference database for metagenomic and metabolomic data, we provide a framework for incorporating additional studies, enabling the expansion of existing knowledge in the form of an evolving community resource. We demonstrate the utility of this database by testing the hypothesis that every microbe and metabolite is everywhere but the environment selects. Our results show that metabolite diversity exhibits turnover and nestedness related to both microbial communities and the environment, whereas the relative abundances of microbially related metabolites vary and co-occur with specific microbial consortia in a habitat-specific manner. We additionally show the power of certain chemistry, in particular terpenoids, in distinguishing Earth's environments (for example, terrestrial plant surfaces and soils, freshwater and marine animal stool), as well as that of certain microbes including Conexibacter woesei (terrestrial soils), Haloquadratum walsbyi (marine deposits) and Pantoea dispersa (terrestrial plant detritus). This Resource provides insight into the taxa and metabolites within microbial communities from diverse habitats across Earth, informing both microbial and chemical ecology, and provides a foundation and methods for multi-omics microbiome studies of hosts and the environment.
© 2022. The Author(s).
Conflict of interest statement
S.B. and K.D. are co-founders of Bright Giant GmbH, which implements some of the tools used for metabolite annotation here (that is, SIRIUS, CSI-FingerID+CANOPUS). The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
