

IL-6/STAT3 signaling in tumor cells restricts the expression of frameshift-derived neoantigens by SMG1 induction
- PMID: 36443756
- PMCID: PMC9703761
- DOI: 10.1186/s12943-022-01679-6
IL-6/STAT3 signaling in tumor cells restricts the expression of frameshift-derived neoantigens by SMG1 induction
Abstract
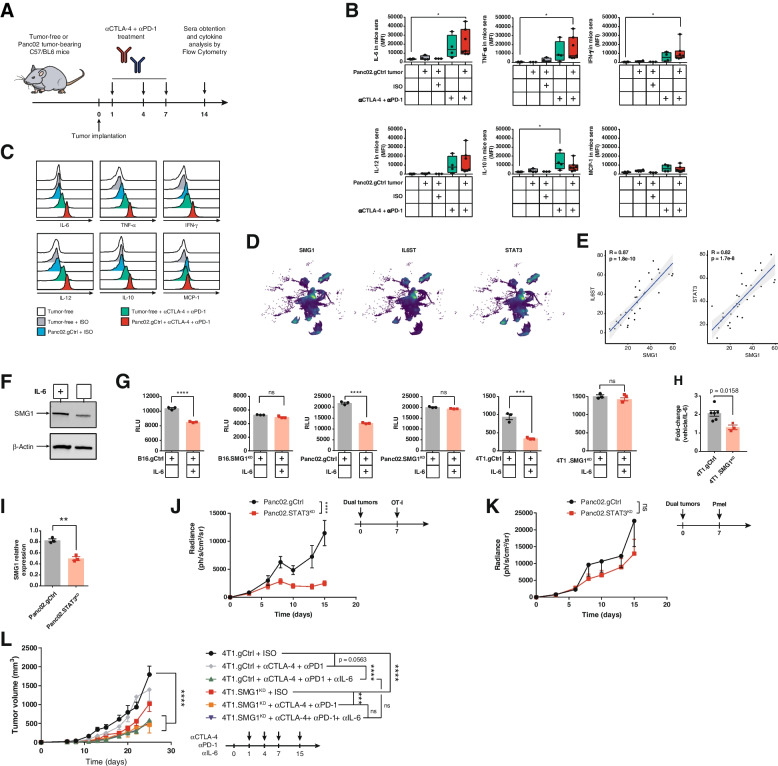
Background: The quality and quantity of tumor neoantigens derived from tumor mutations determines the fate of the immune response in cancer. Frameshift mutations elicit better tumor neoantigens, especially when they are not targeted by nonsense-mediated mRNA decay (NMD). For tumor progression, malignant cells need to counteract the immune response including the silencing of immunodominant neoantigens (antigen immunoediting) and promoting an immunosuppressive tumor microenvironment. Although NMD inhibition has been reported to induce tumor immunity and increase the expression of cryptic neoantigens, the possibility that NMD activity could be modulated by immune forces operating in the tumor microenvironment as a new immunoediting mechanism has not been addressed.
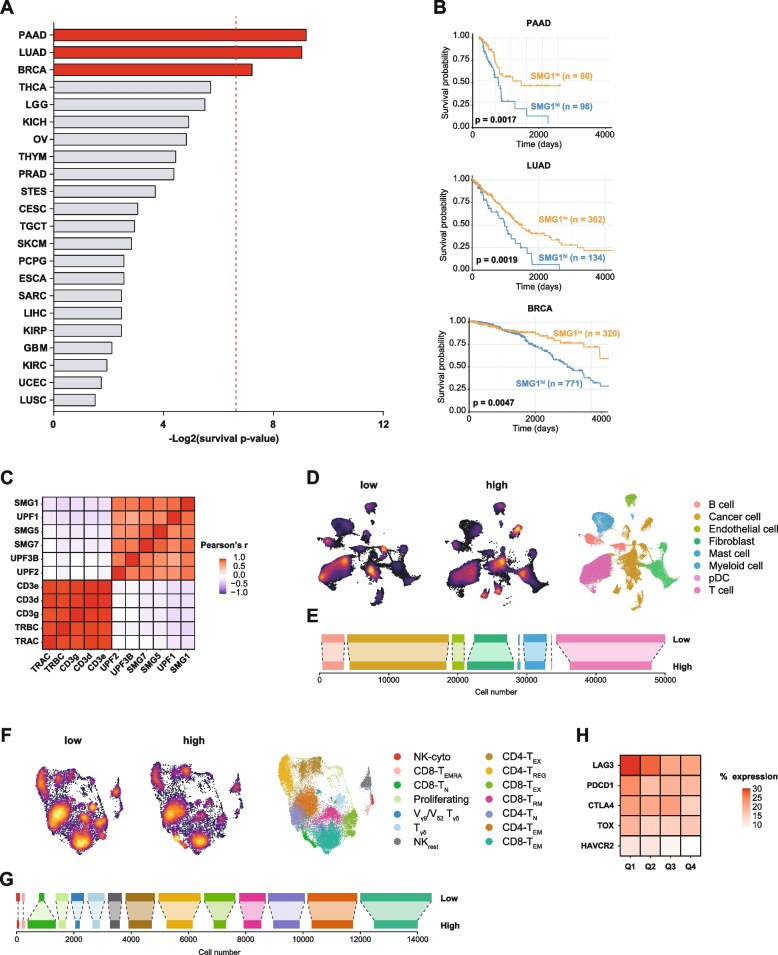
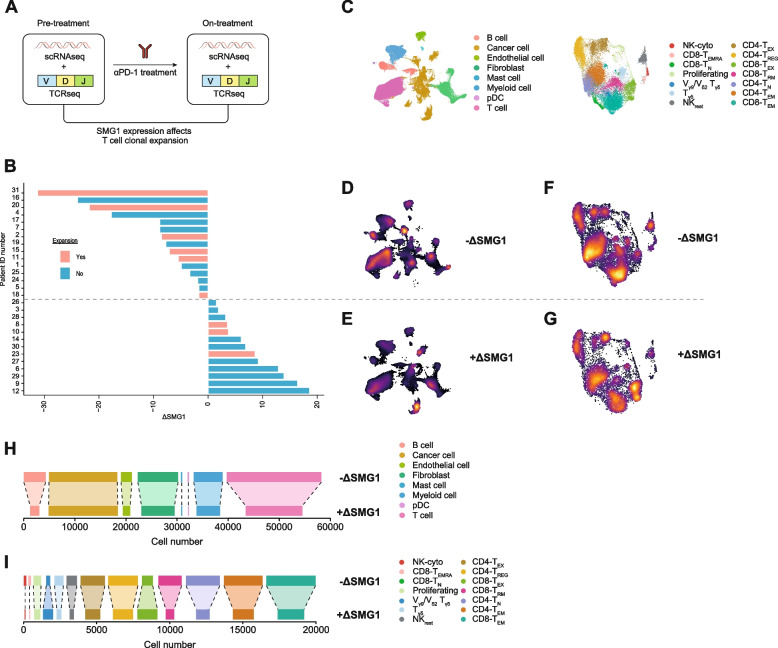
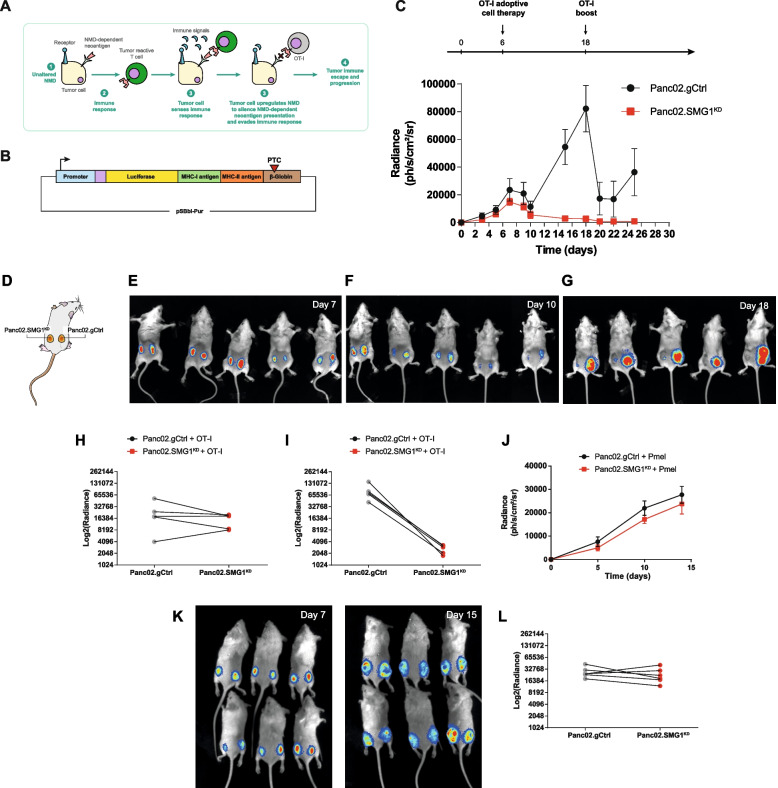
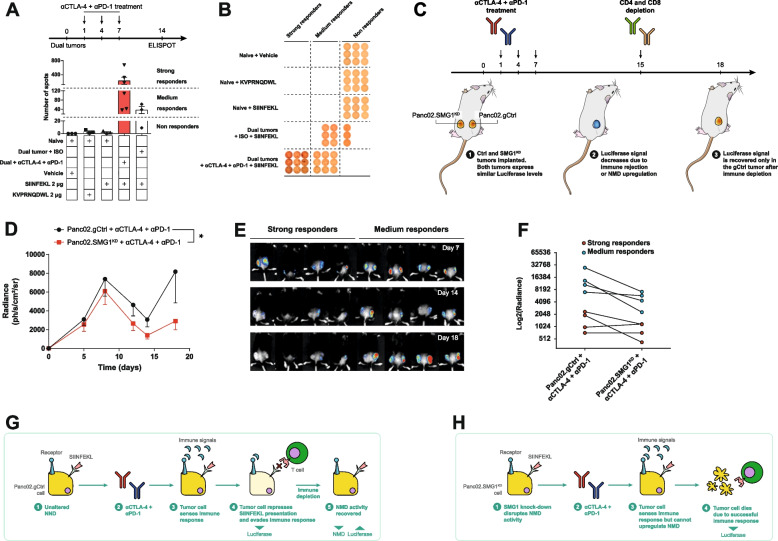
Methods: We study the effect of SMG1 expression (main kinase that initiates NMD) in the survival and the nature of the tumor immune infiltration using TCGA RNAseq and scRNAseq datasets of breast, lung and pancreatic cancer. Different murine tumor models were used to corroborate the antitumor immune dependencies of NMD. We evaluate whether changes of SMG1 expression in malignant cells impact the immune response elicited by cancer immunotherapy. To determine how NMD fluctuates in malignant cells we generated a luciferase reporter system to track NMD activity in vivo under different immune conditions. Cytokine screening, in silico studies and functional assays were conducted to determine the regulation of SMG1 via IL-6/STAT3 signaling.
Results: IL-6/STAT3 signaling induces SMG1, which limits the expression of potent frameshift neoantigens that are under NMD control compromising the outcome of the immune response.
Conclusion: We revealed a new neoantigen immunoediting mechanism regulated by immune forces (IL-6/STAT3 signaling) responsible for silencing otherwise potent frameshift mutation-derived neoantigens.
Keywords: Cancer immunotherapy; Immunoediting; NMD; Neoantigens; Tumor immunity.
© 2022. The Author(s).
Conflict of interest statement
Authors declare that they have no competing interests.
Figures


References
-
- Korman AJ, Garrett-Thomson SC, Lonberg N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat Rev Drug Discov. 2021. - PubMed
-
- Yap TA, Parkes EE, Peng W, Moyers JT, Curran MA, Tawbi HA. Development of immunotherapy combination strategies in cancer. Cancer Discov. 2021;11:1368–1397. doi: 10.1158/2159-8290.CD-20-1209. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
