

Noninvasive prenatal screening for cystic fibrosis using circulating trophoblasts: Detection of the 50 most common disease-causing variants
- PMID: 36447355
- PMCID: PMC10107343
- DOI: 10.1002/pd.6276
Noninvasive prenatal screening for cystic fibrosis using circulating trophoblasts: Detection of the 50 most common disease-causing variants
Abstract
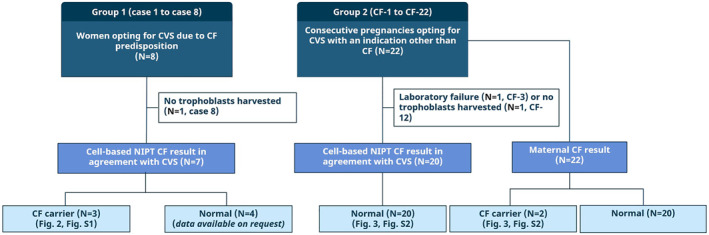
Objectives: Cystic fibrosis (CF) is one of the most common severe autosomal recessive disorders. Prenatal or preconception CF screening is offered in some countries. A maternal blood sample in early pregnancy can provide circulating trophoblasts and offers a DNA source for genetic analysis of both the mother and the fetus. This study aimed to develop a cell-based noninvasive prenatal test (NIPT) to screen for the 50 most common CF variants.
Methods: Blood samples were collected from 30 pregnancies undergoing invasive diagnostics and circulating trophoblasts were harvested in 27. Cystic fibrosis testing was conducted using two different methods: by fragment length analysis and by our newly developed NGS-based CF analysis.
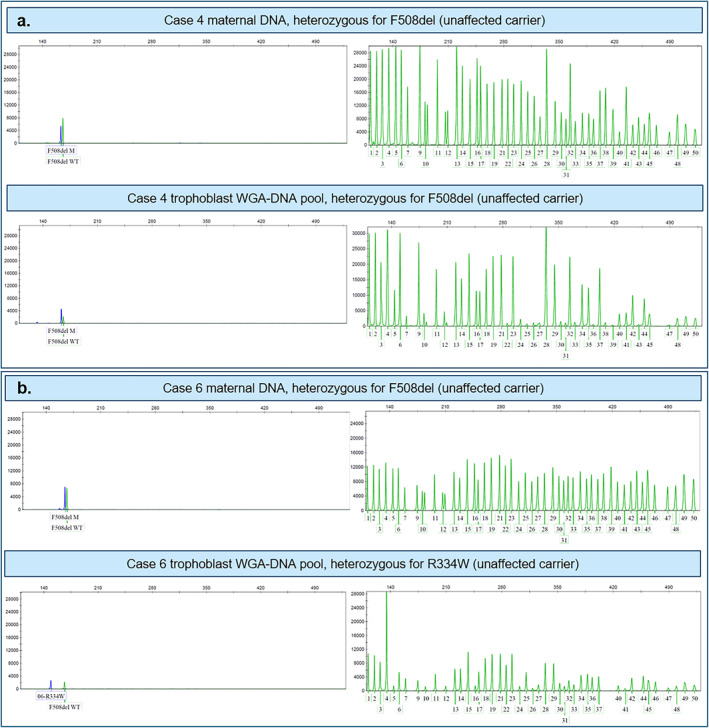
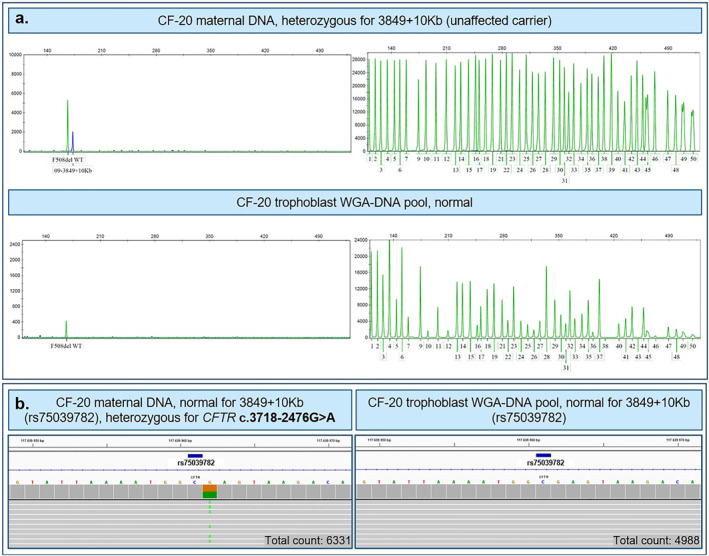
Results: In all 27 cases, cell-based NIPT provided a result using both methods in agreement with the invasive test result.
Conclusion: This study shows that cell-based NIPT for CF screening provides a reliable result without the need for partner- and proband samples.
© 2022 The Authors. Prenatal Diagnosis published by John Wiley & Sons Ltd.
Conflict of interest statement
Line Dahl Jeppesen, Lotte Hatt, Jakob Hedegaard, Ripudaman Singh, and PS are all employed by ARCEDI, a Danish biotech company that holds the patented technology for enrichment of circulating trophoblasts used in this study. Anders Sune Pedersen and Michael Knudsen are employed as consultants by ARCEDI. Dorte Launholt Lildballe, Christian Liebst Frisk Toft, and Ida Vogel have no conflicts of interest and do not receive any funding by ARCEDI.
Figures
References
-
- Hospital of Sick Children . Cystic Fibrosis Mutation Database [Internet]. [cited 2022 Mar 10]. http://www.genet.sickkids.on.ca
-
- Romero S, Biggio JR, Saller DN, Giardine R. Committee Opinion Number 432. Vol 486. Committee Opinion Number; 2005.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
