

Generation and multiomic profiling of a TP53/CDKN2A double-knockout gastroesophageal junction organoid model
- PMID: 36449602
- PMCID: PMC10026384
- DOI: 10.1126/scitranslmed.abq6146
Generation and multiomic profiling of a TP53/CDKN2A double-knockout gastroesophageal junction organoid model
Abstract
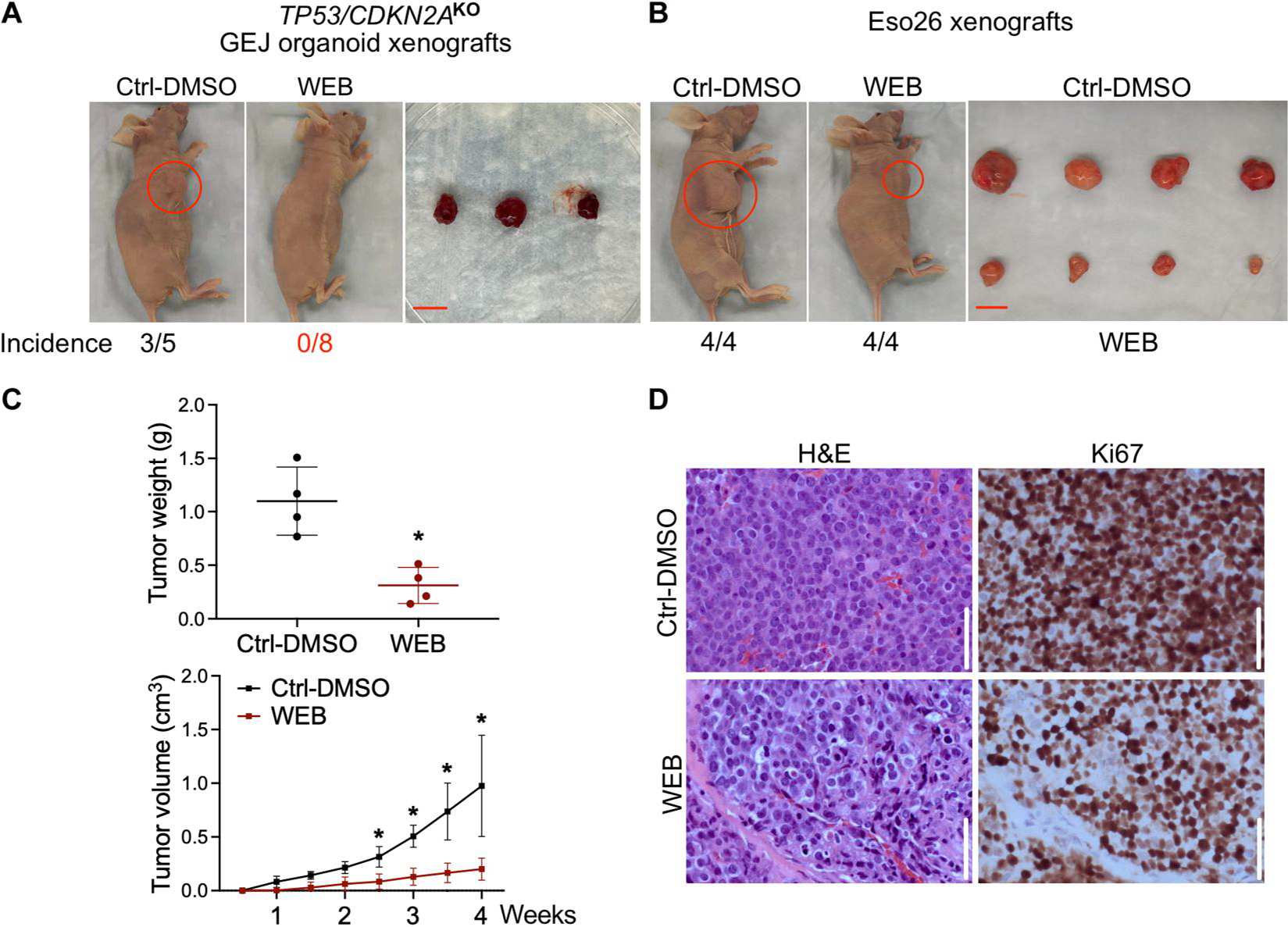
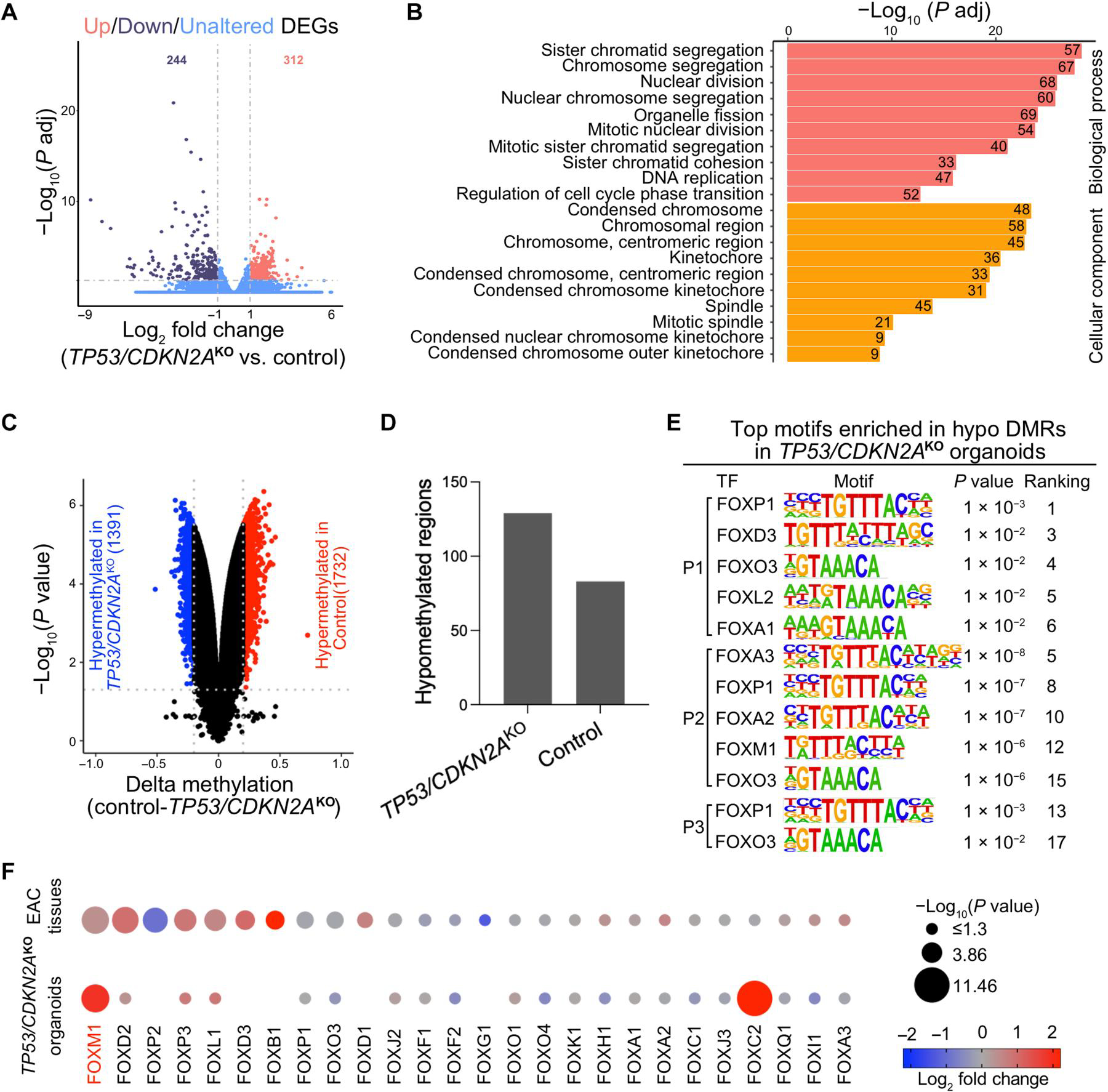
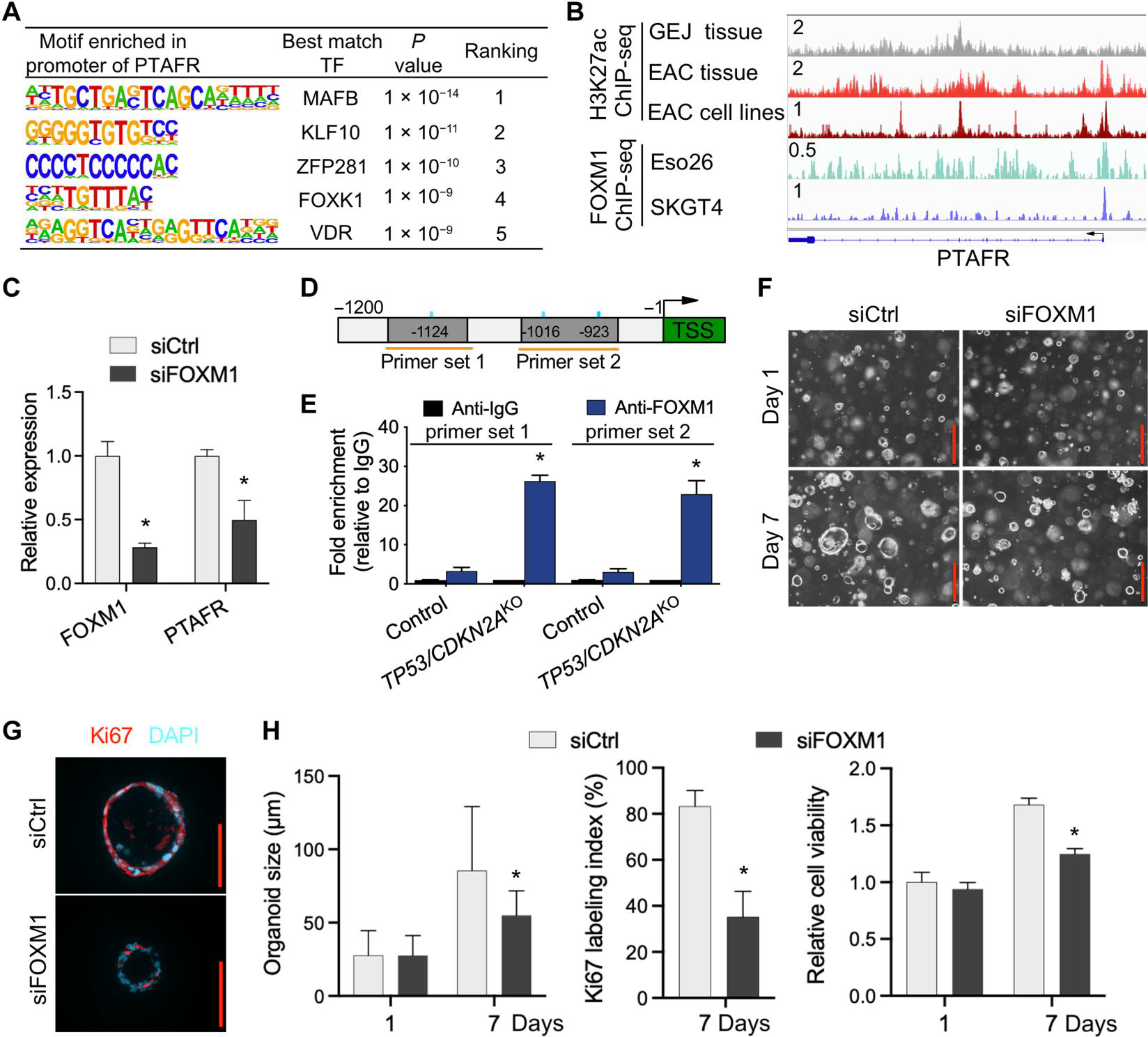
Inactivation of the tumor suppressor genes tumor protein p53 (TP53) and cyclin-dependent kinase inhibitor 2A (CDKN2A) occurs early during gastroesophageal junction (GEJ) tumorigenesis. However, because of a paucity of GEJ-specific disease models, cancer-promoting consequences of TP53 and CDKN2A inactivation at the GEJ have not been characterized. Here, we report the development of a wild-type primary human GEJ organoid model and a CRISPR-edited transformed GEJ organoid model. CRISPR-Cas9-mediated TP53 and CDKN2A knockout (TP53/CDKN2AKO) in GEJ organoids induced morphologic dysplasia and proneoplastic features in vitro and tumor formation in vivo. Lipidomic profiling identified several platelet-activating factors (PTAFs) among the most up-regulated lipids in CRISPR-edited organoids. PTAF/PTAF receptor (PTAFR) abrogation by siRNA knockdown or a pharmacologic inhibitor (WEB2086) reduced proliferation and other proneoplastic features of TP53/CDKN2AKO GEJ organoids in vitro and tumor formation in vivo. In addition, murine xenografts of Eso26, an established human esophageal adenocarcinoma cell line, were suppressed by WEB2086. Mechanistically, TP53/CDKN2A dual inactivation disrupted both the transcriptome and the DNA methylome, likely mediated by key transcription factors, particularly forkhead box M1 (FOXM1). FOXM1 activated PTAFR transcription by binding to the PTAFR promoter, further amplifying the PTAF-PTAFR pathway. Together, these studies established a robust model system for investigating early GEJ neoplastic events, identified crucial metabolic and epigenomic changes occurring during GEJ model tumorigenesis, and revealed a potential cancer therapeutic strategy. This work provides insights into proneoplastic mechanisms associated with TP53/CDKN2A inactivation in early GEJ neoplasia, which may facilitate early diagnosis and prevention of GEJ neoplasms.
Conflict of interest statement
Figures
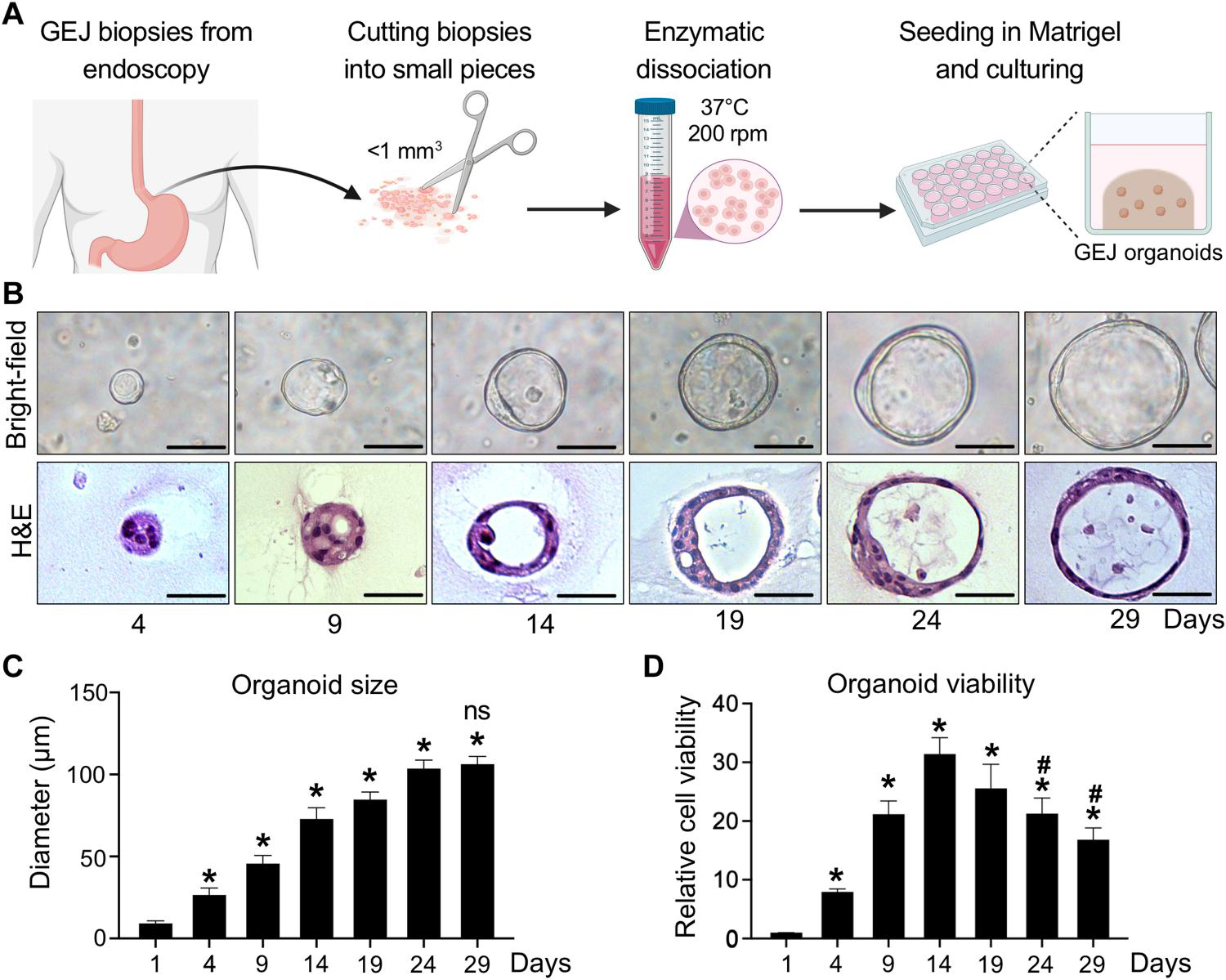
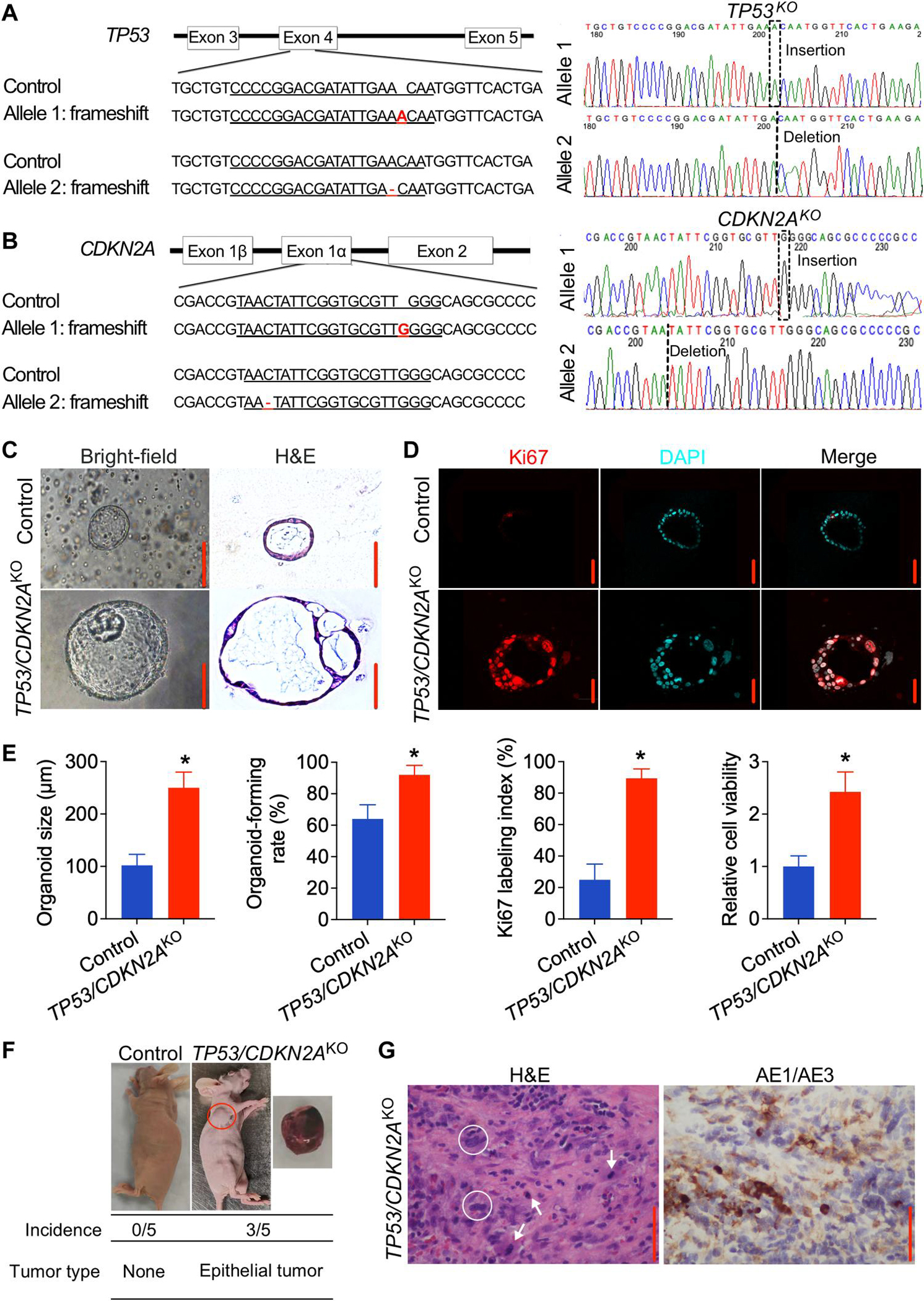
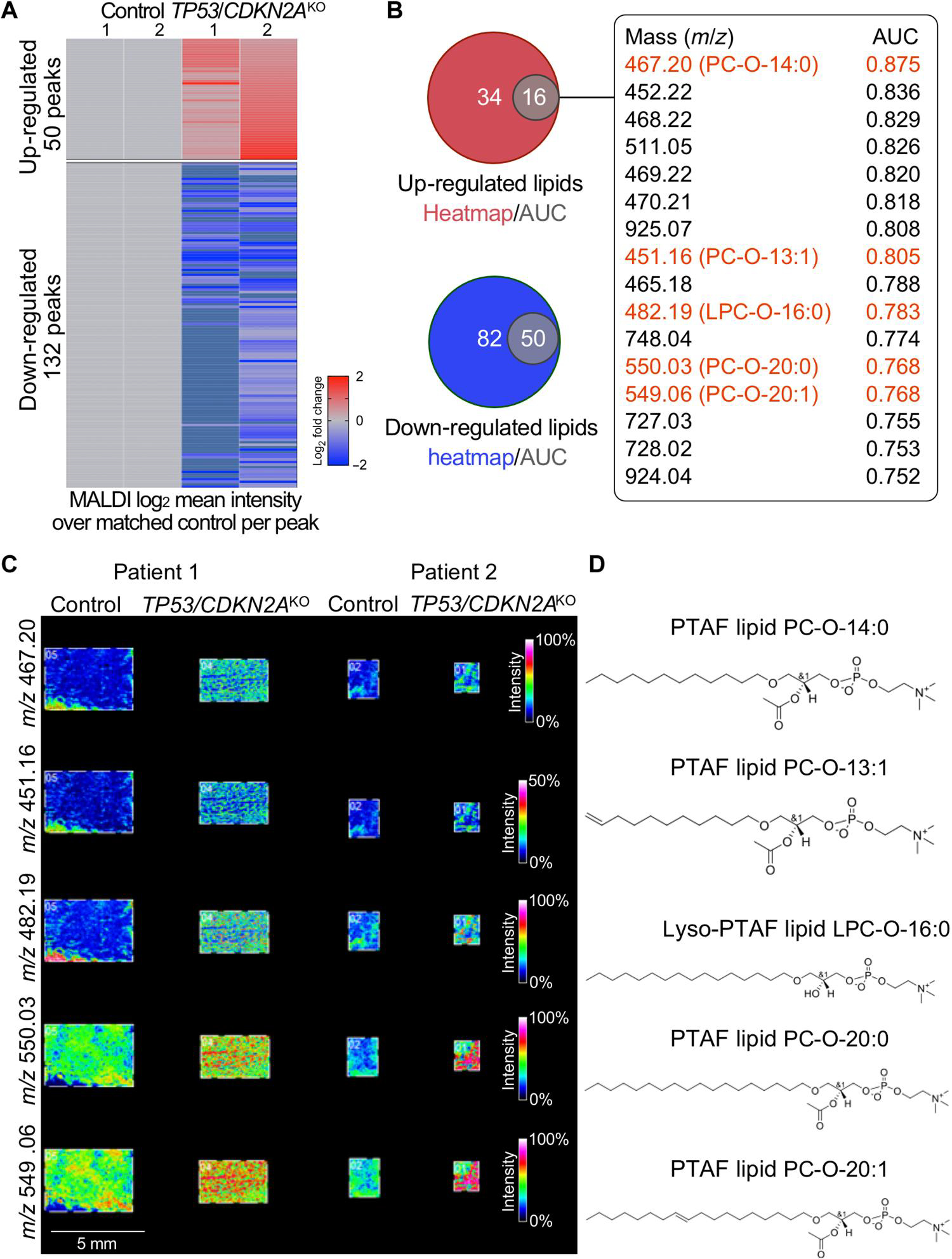
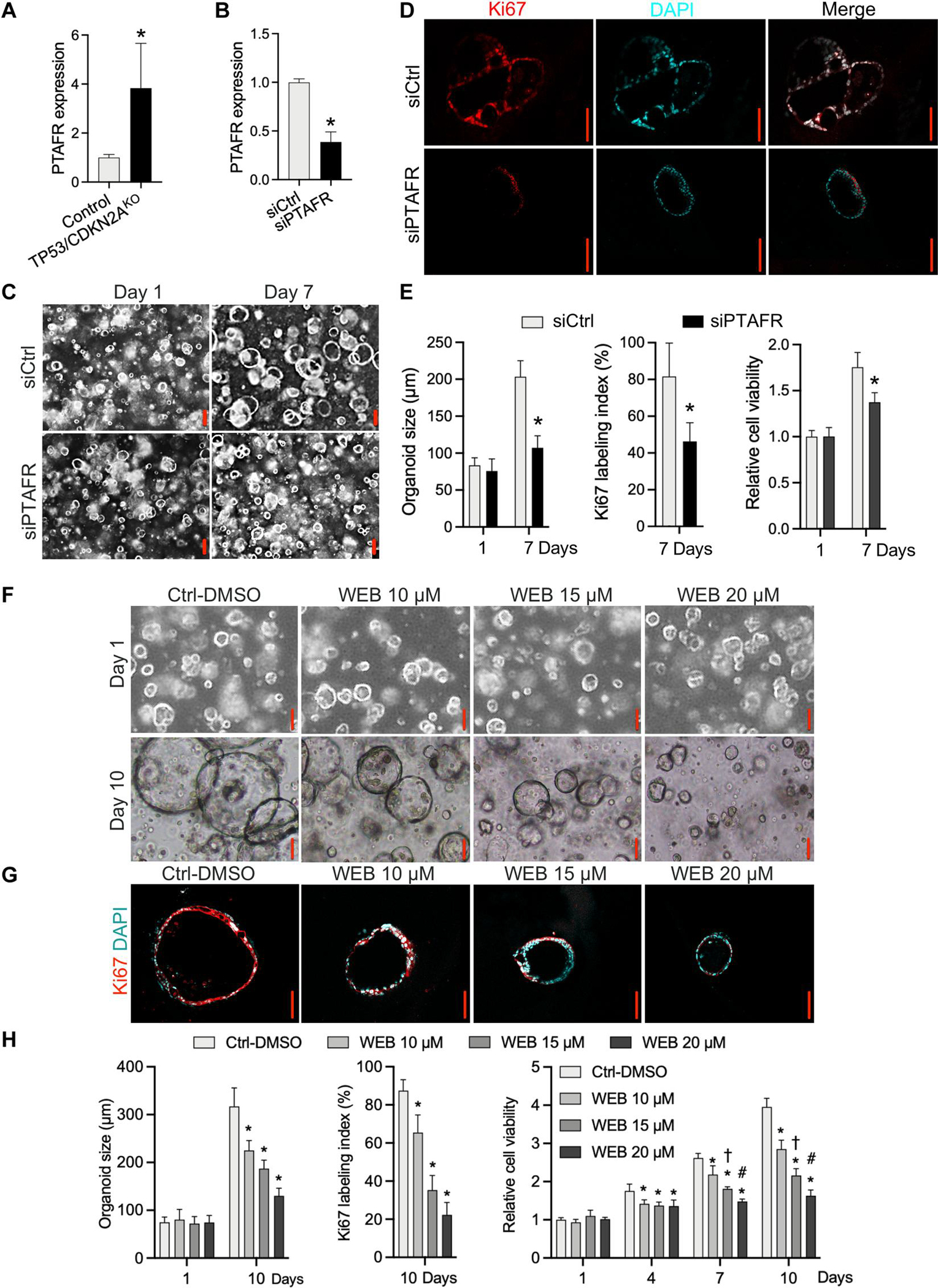
References
-
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F, Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021). - PubMed
-
- Bartel M, Brahmbhatt B, Bhurwal A, Incidence of gastroesophageal junction cancer continues to rise: Analysis of Surveillance, Epidemiology, and End Results (SEER) database. J. Clin. Orthod. 37, 40 (2019).
-
- Snaebjornsson MT, Janaki-Raman S, Schulze A, Greasing the wheels of the cancer machine: The role of lipid metabolism in cancer. Cell Metab. 31, 62–76 (2020). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
