

Ras drives malignancy through stem cell crosstalk with the microenvironment
- PMID: 36450983
- PMCID: PMC9750880
- DOI: 10.1038/s41586-022-05475-6
Ras drives malignancy through stem cell crosstalk with the microenvironment
Abstract
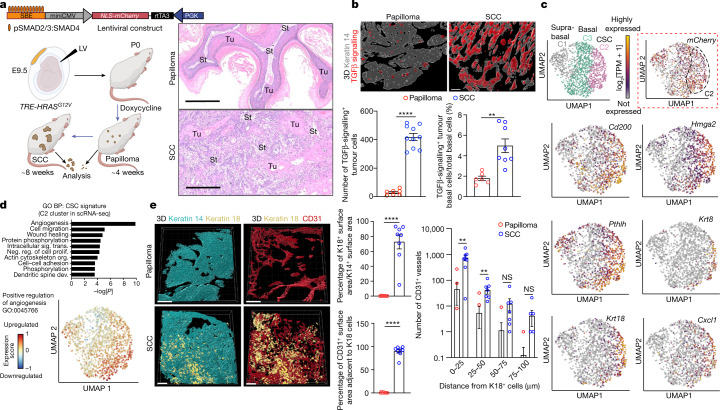
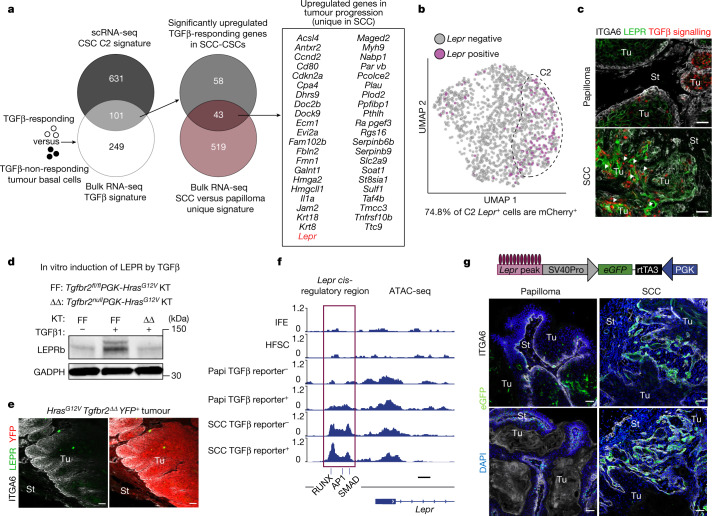
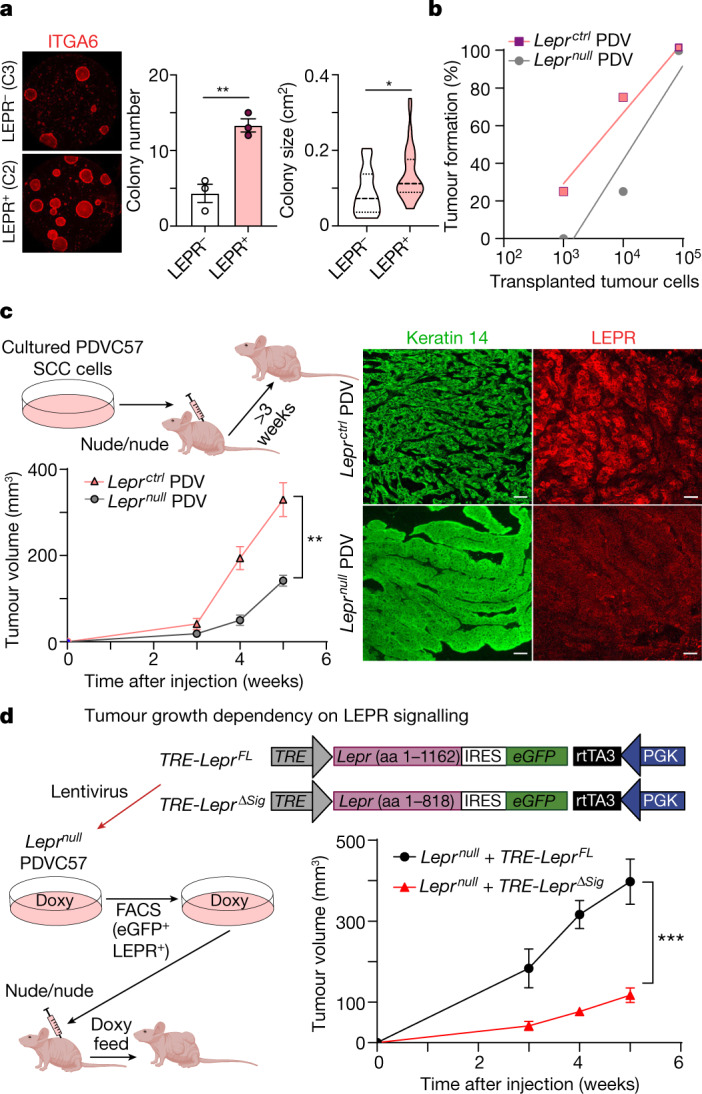
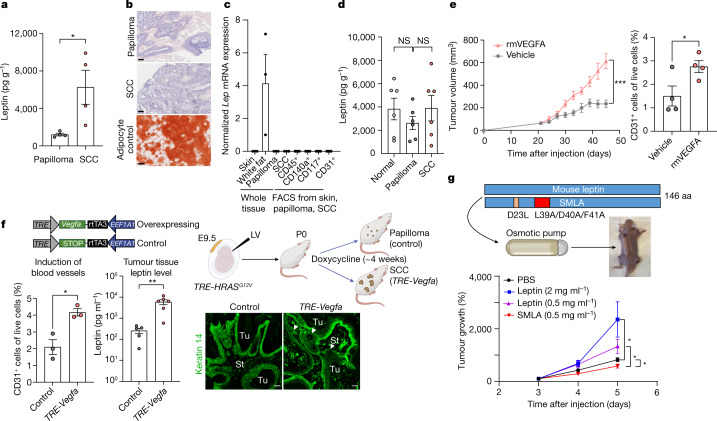
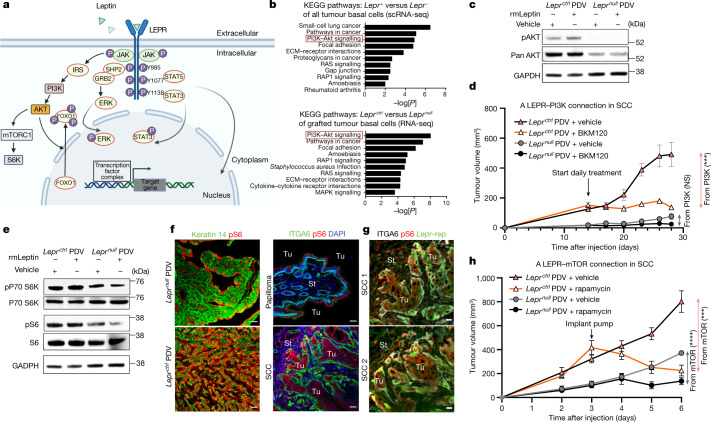
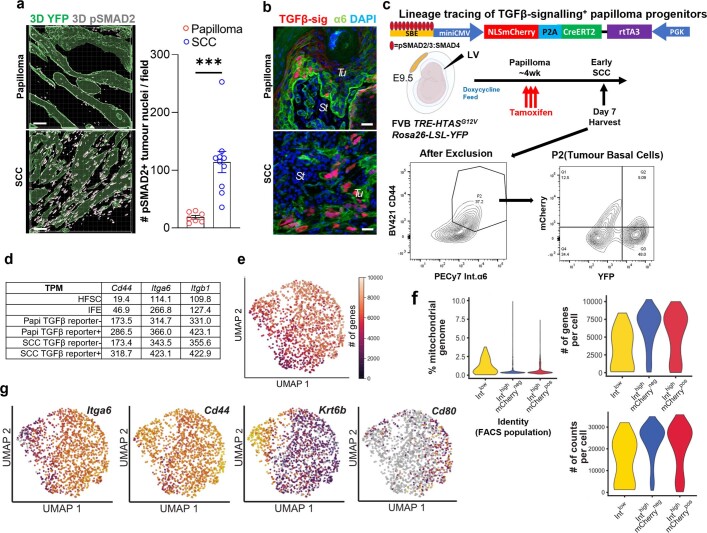
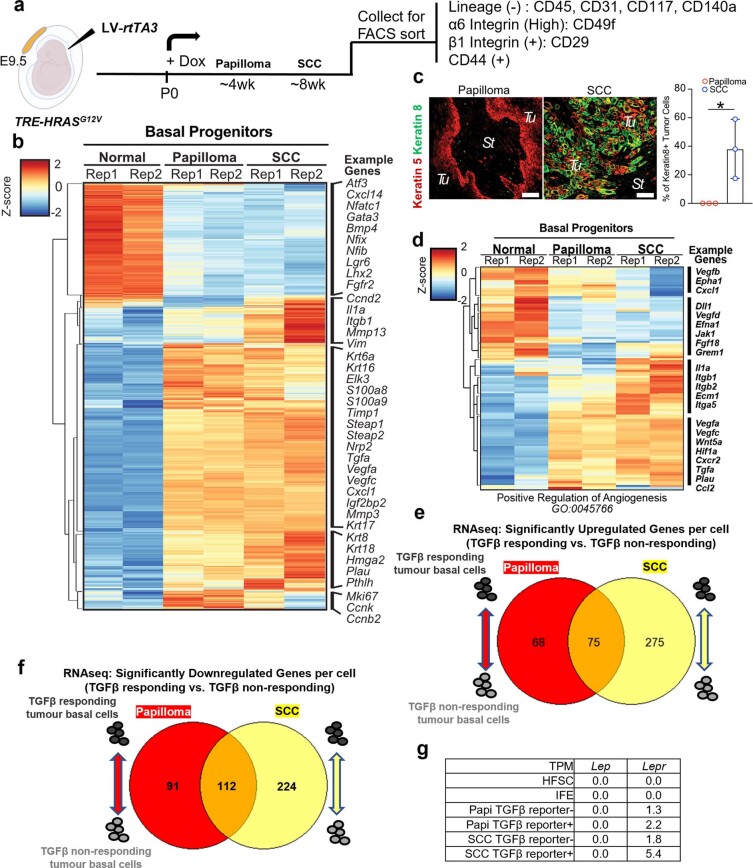
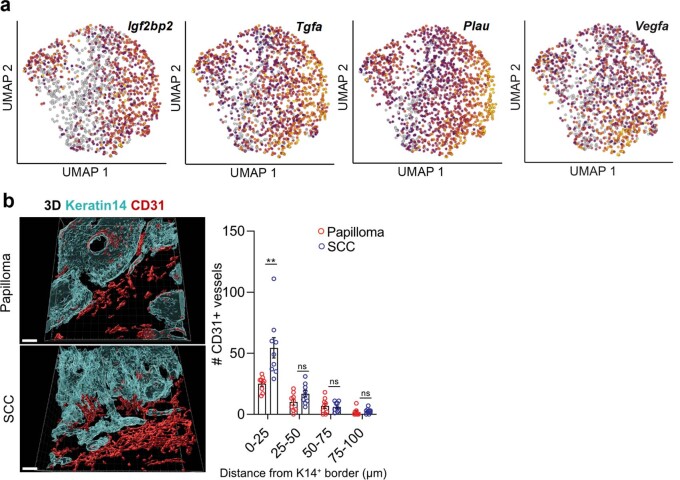
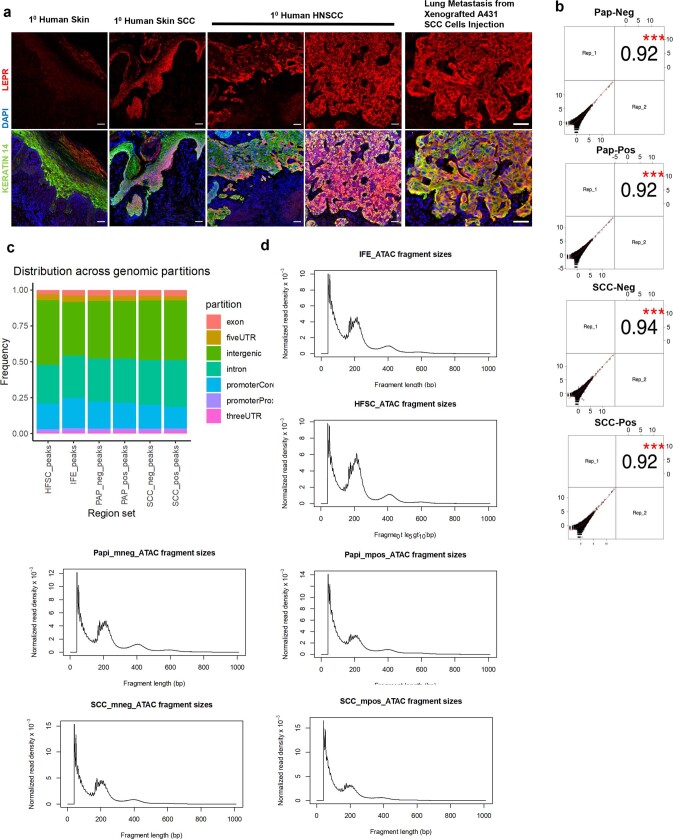
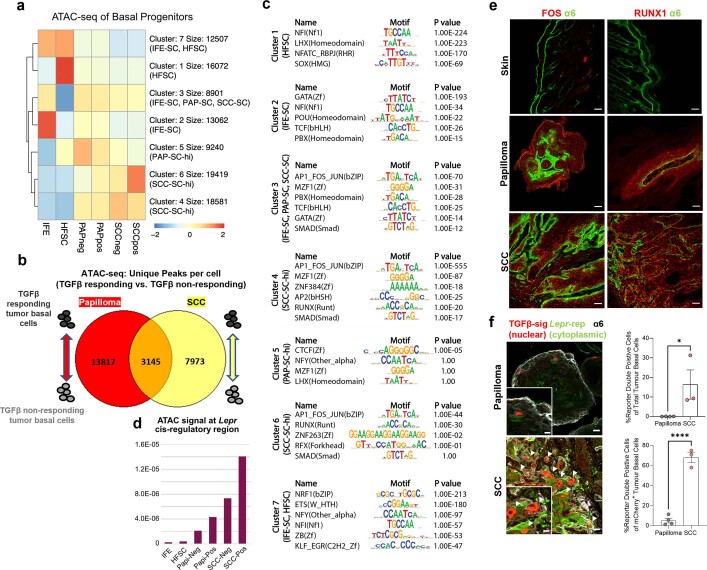
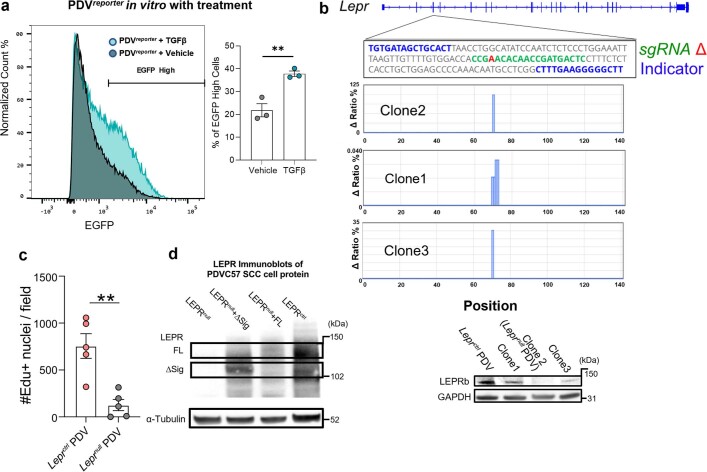
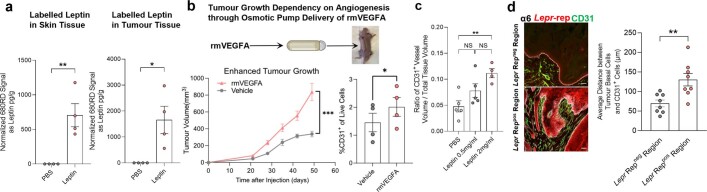
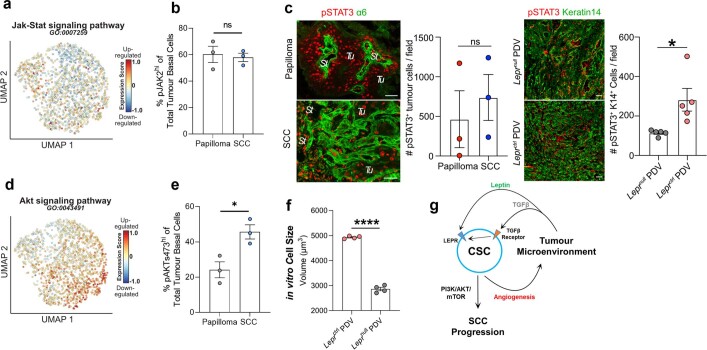
Squamous cell carcinomas are triggered by marked elevation of RAS-MAPK signalling and progression from benign papilloma to invasive malignancy1-4. At tumour-stromal interfaces, a subset of tumour-initiating progenitors, the cancer stem cells, obtain increased resistance to chemotherapy and immunotherapy along this pathway5,6. The distribution and changes in cancer stem cells during progression from a benign state to invasive squamous cell carcinoma remain unclear. Here we show in mice that, after oncogenic RAS activation, cancer stem cells rewire their gene expression program and trigger self-propelling, aberrant signalling crosstalk with their tissue microenvironment that drives their malignant progression. The non-genetic, dynamic cascade of intercellular exchanges involves downstream pathways that are often mutated in advanced metastatic squamous cell carcinomas with high mutational burden7. Coupling our clonal skin HRASG12V mouse model with single-cell transcriptomics, chromatin landscaping, lentiviral reporters and lineage tracing, we show that aberrant crosstalk between cancer stem cells and their microenvironment triggers angiogenesis and TGFβ signalling, creating conditions that are conducive for hijacking leptin and leptin receptor signalling, which in turn launches downstream phosphoinositide 3-kinase (PI3K)-AKT-mTOR signalling during the benign-to-malignant transition. By functionally examining each step in this pathway, we reveal how dynamic temporal crosstalk with the microenvironment orchestrated by the stem cells profoundly fuels this path to malignancy. These insights suggest broad implications for cancer therapeutics.
© 2022. The Author(s).
Conflict of interest statement
E.F. has served on the scientific advisory boards of L’Oreal and Arsenal Biosciences. The other authors declare no competing interests.
Figures
Comment in
-
Leptin fuels non-genetic skin tumour progression.Nat Rev Cancer. 2023 Feb;23(2):56. doi: 10.1038/s41568-023-00548-8. Nat Rev Cancer. 2023. PMID: 36631549 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
