

AKR1C3-dependent lipid droplet formation confers hepatocellular carcinoma cell adaptability to targeted therapy
- PMID: 36451864
- PMCID: PMC9706585
- DOI: 10.7150/thno.74974
AKR1C3-dependent lipid droplet formation confers hepatocellular carcinoma cell adaptability to targeted therapy
Abstract
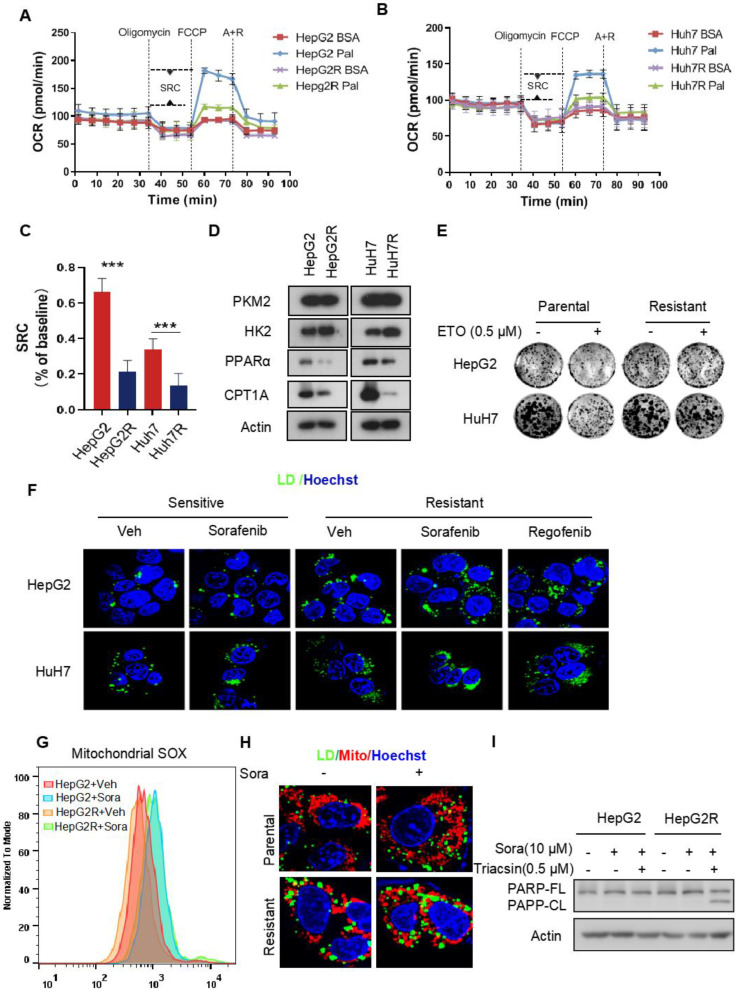
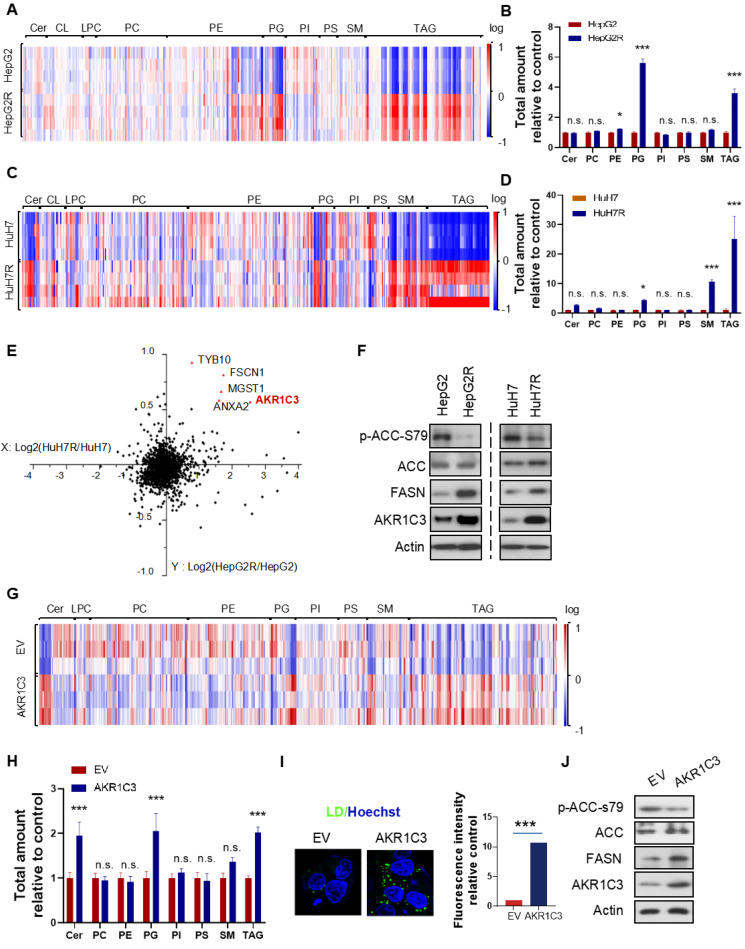
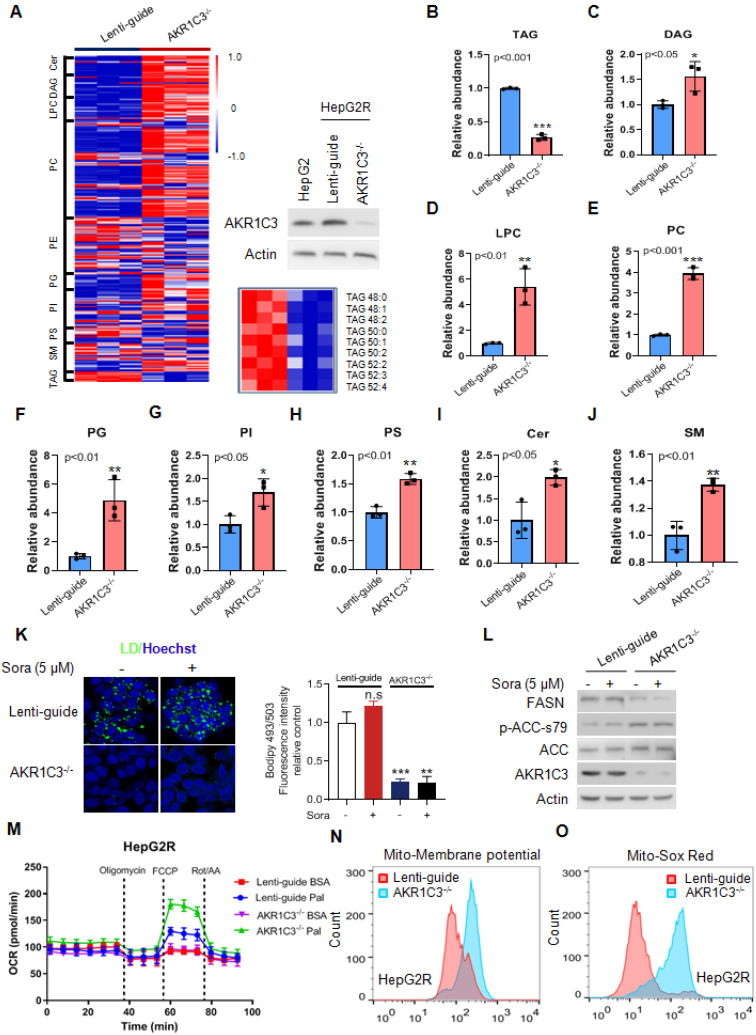
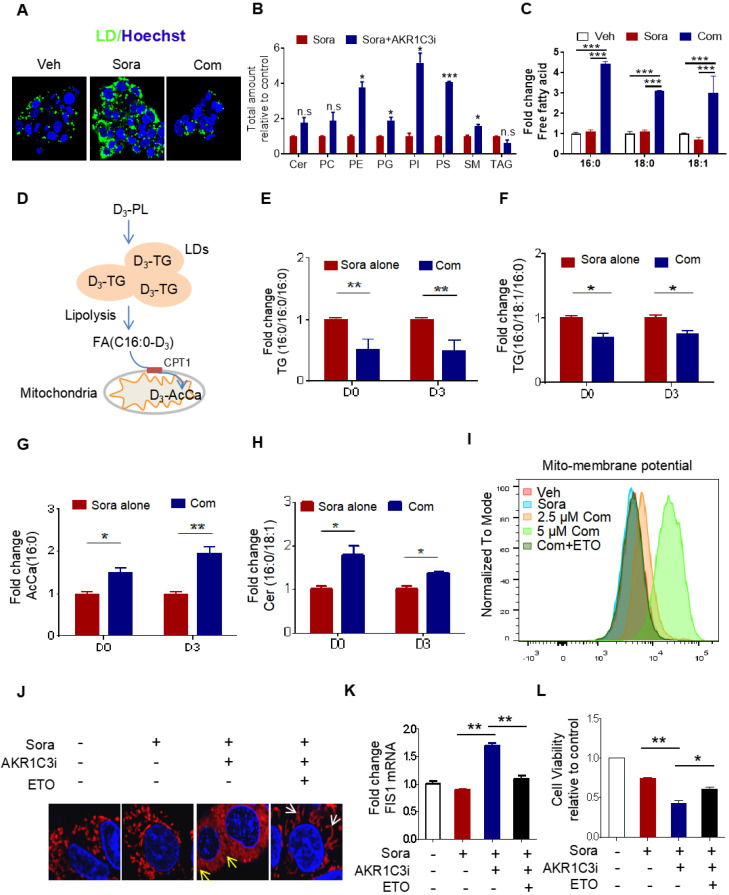
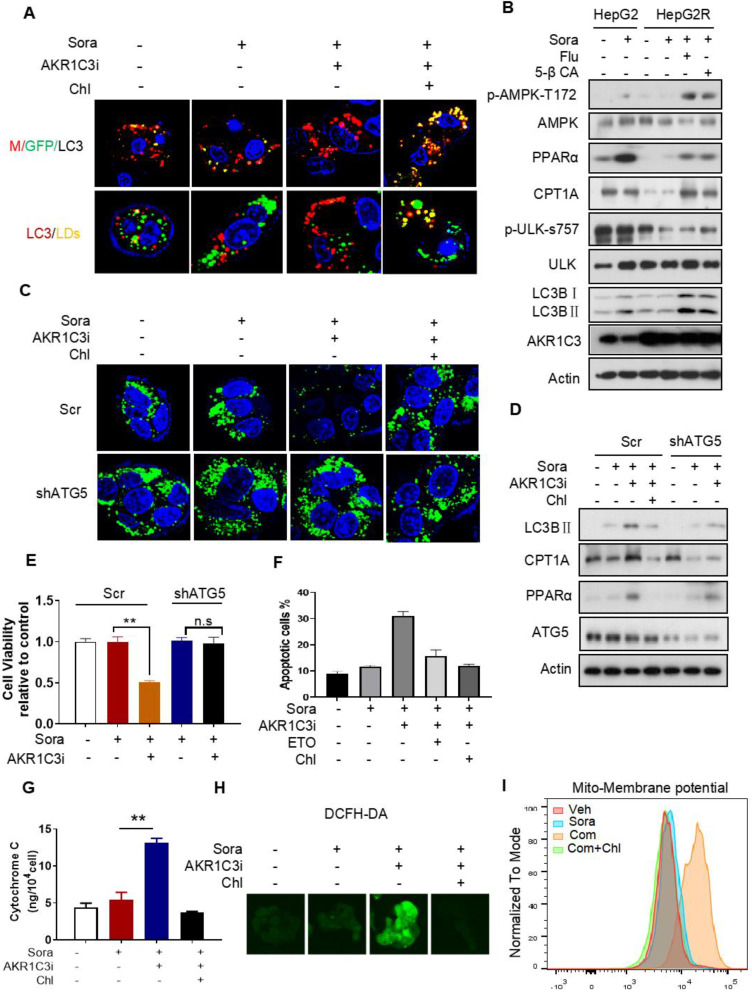
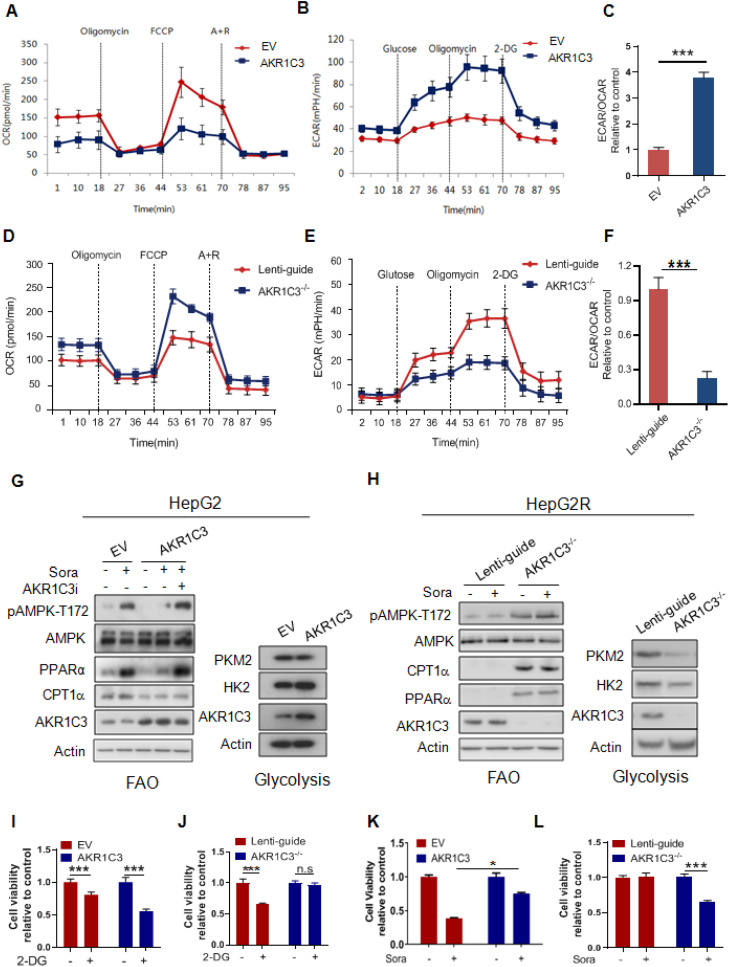
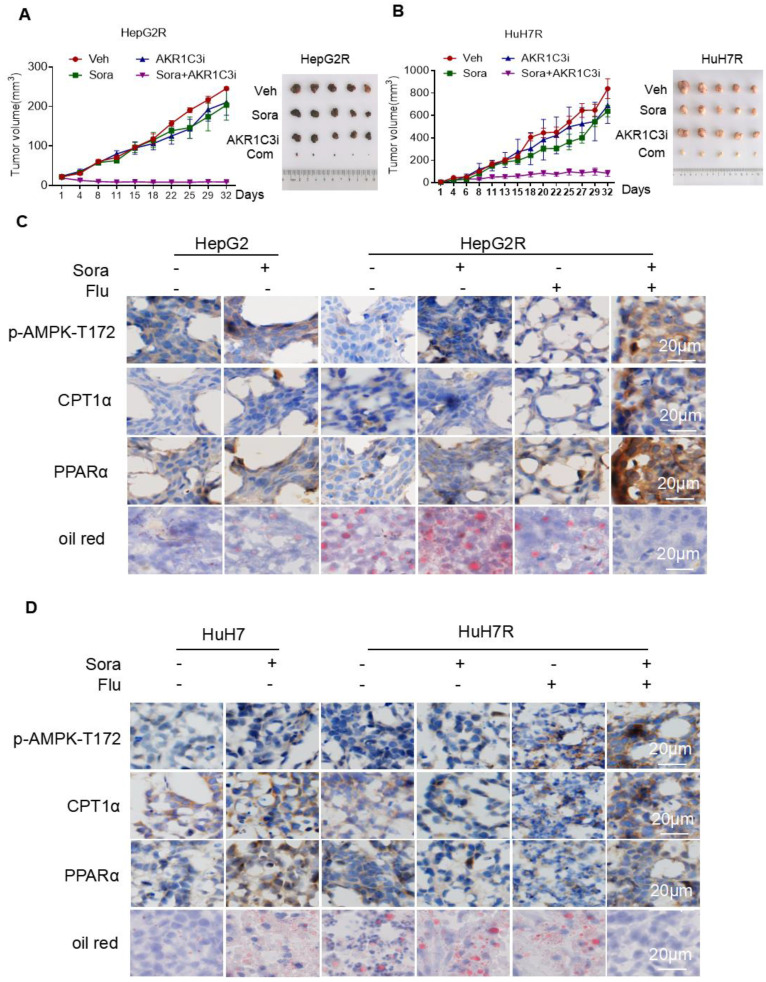
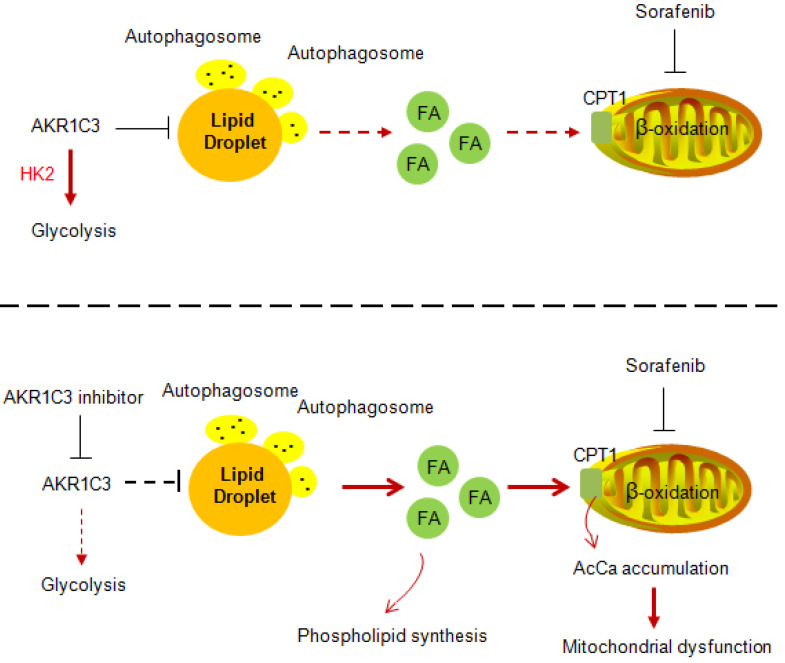
Rationale: Increased lipid droplet (LD) formation has been linked to tumor metastasis, stemness, and chemoresistance in various types of cancer. Here, we revealed that LD formation is critical for the adaptation to sorafenib in hepatocellular carcinoma (HCC) cells. We aim to investigate the LD function and its regulatory mechanisms in HCC. Methods: The key proteins responsible for LD formation were screened by both metabolomics and proteomics in sorafenib-resistant HCC cells and further validated by immunoblotting and immunofluorescence staining. Biological function of AKR1C3 was evaluated by CRISPR/Cas9-based gene editing. Isotopic tracing analysis with deuterium3-labeled palmitate or carbon13-labeled glucose was conducted to investigate fatty acid (FA) and glucose carbon flux. Seahorse analysis was performed to assess the glycolytic flux and mitochondrial function. Selective AKR1C3 inhibitors were used to evaluate the effect of AKR1C3 inhibition on HCC tumor growth and induction of autophagy. Results: We found that long-term sorafenib treatment impairs fatty acid oxidation (FAO), leading to LD accumulation in HCC cells. Using multi-omics analysis in cultured HCC cells, we identified that aldo-keto reductase AKR1C3 is responsible for LD accumulation in HCC. Genetic loss of AKR1C3 fully depletes LD contents, navigating FA flux to phospholipids, sphingolipids, and mitochondria. Furthermore, we found that AKR1C3-dependent LD accumulation is required for mitigating sorafenib-induced mitochondrial lipotoxicity and dysfunction. Pharmacologic inhibition of AKR1C3 activity instantly induces autophagy-dependent LD catabolism, resulting in mitochondrial fission and apoptosis in sorafenib-resistant HCC clones. Notably, manipulation of AKR1C3 expression is sufficient to drive the metabolic switch between FAO and glycolysis. Conclusions: Our findings revealed that AKR1C3-dependent LD formation is critical for the adaptation to sorafenib in HCC through regulating lipid and energy homeostasis. AKR1C3-dependent LD accumulation protects HCC cells from sorafenib-induced mitochondrial lipotoxicity by regulating lipophagy. Targeting AKR1C3 might be a promising therapeutic strategy for HCC tumors.
Keywords: Lipid droplets; Lipid homeostasis; Lipophagy; Metabolic reprogramming; Mitochondrial dysfunction..
© The author(s).
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
