

Homozygous COQ7 mutation: a new cause of potentially treatable distal hereditary motor neuropathy
- PMID: 36454683
- PMCID: PMC10393394
- DOI: 10.1093/brain/awac453
Homozygous COQ7 mutation: a new cause of potentially treatable distal hereditary motor neuropathy
Abstract
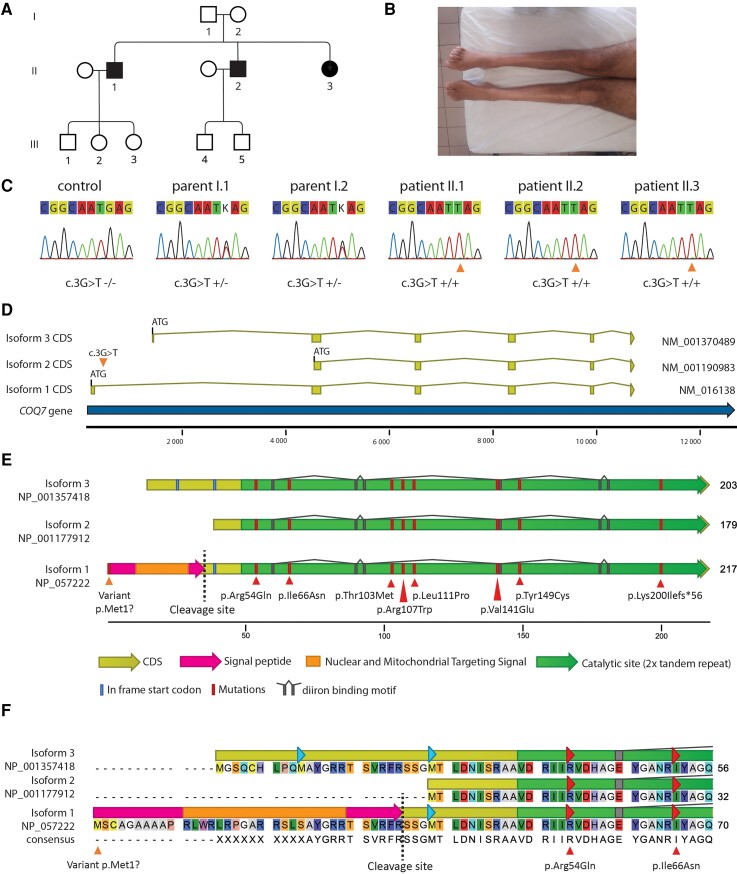
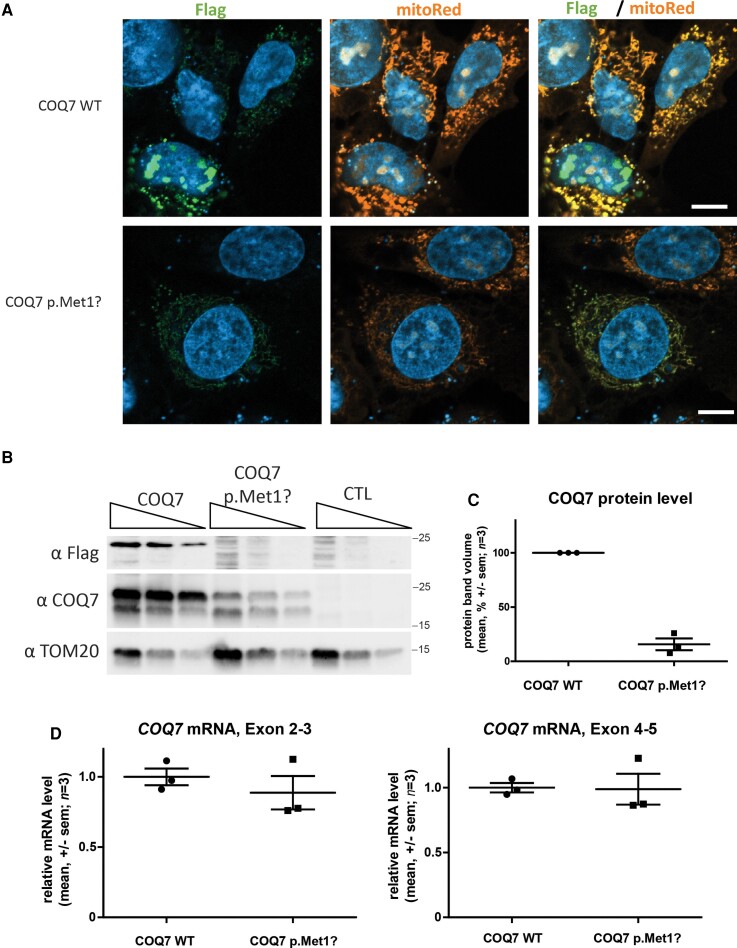
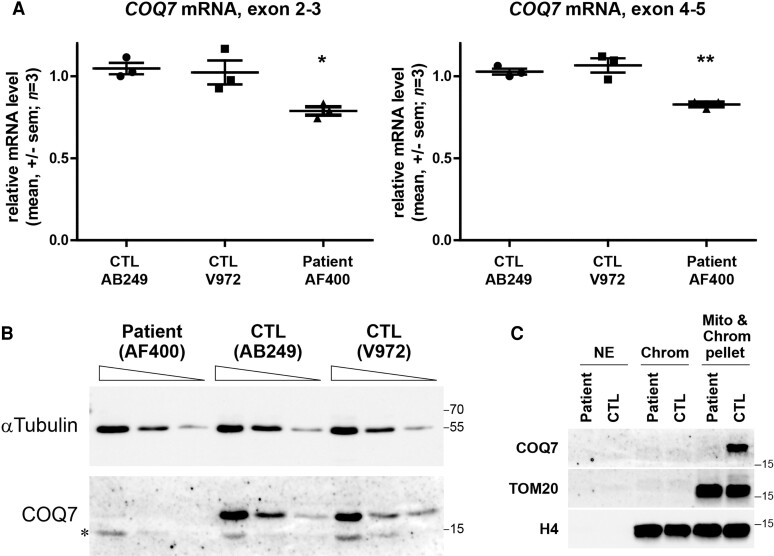
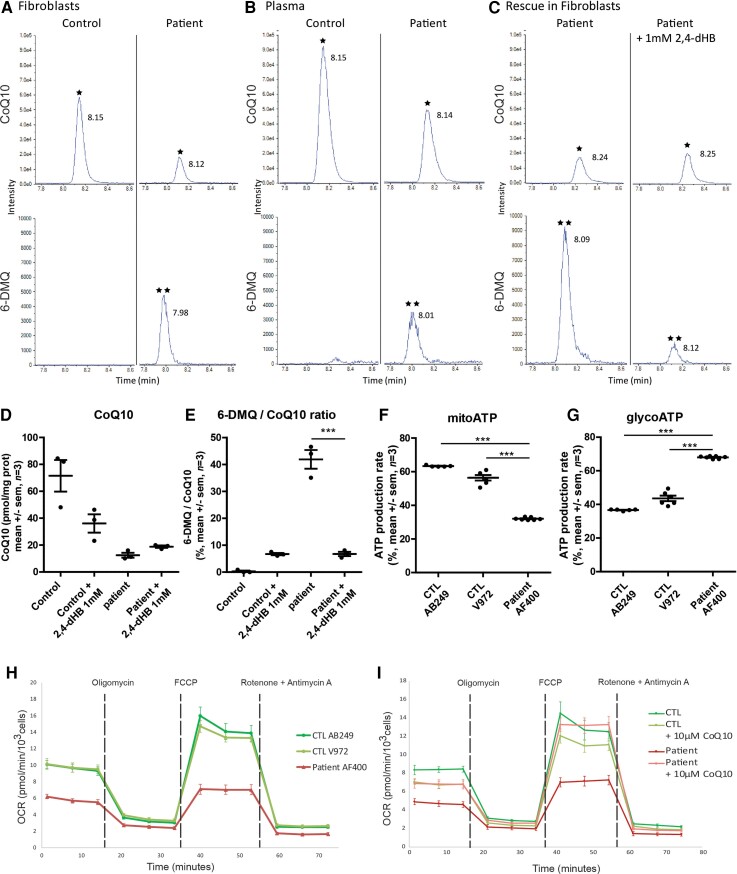
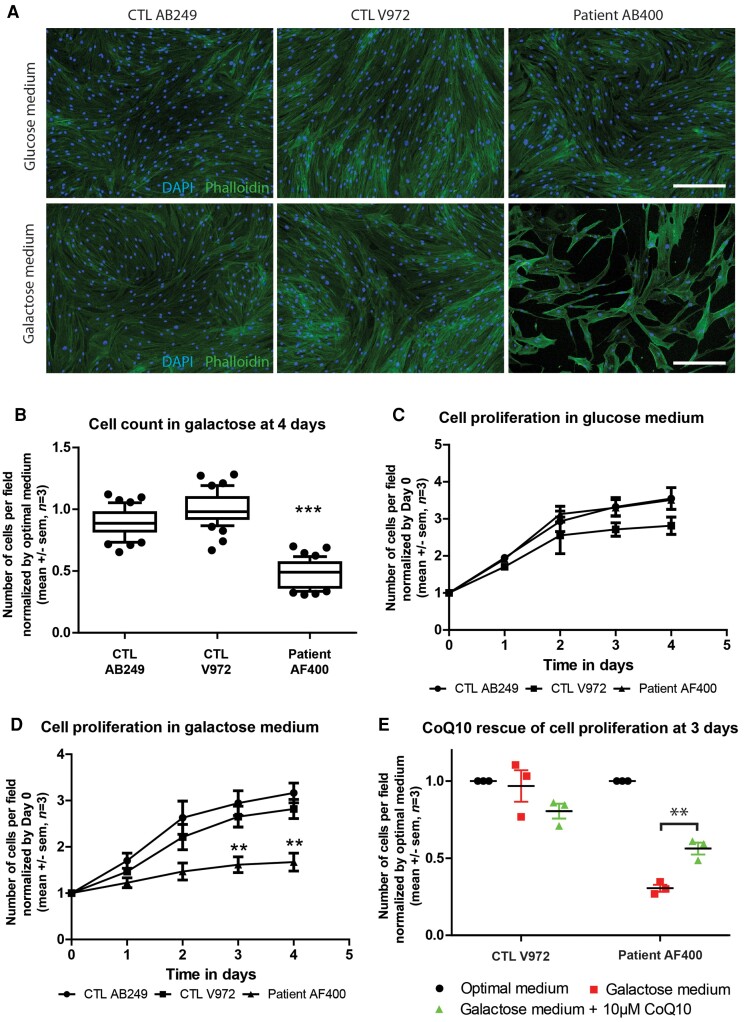
Distal hereditary motor neuropathy represents a group of motor inherited neuropathies leading to distal weakness. We report a family of two brothers and a sister affected by distal hereditary motor neuropathy in whom a homozygous variant c.3G>T (p.1Met?) was identified in the COQ7 gene. This gene encodes a protein required for coenzyme Q10 biosynthesis, a component of the respiratory chain in mitochondria. Mutations of COQ7 were previously associated with severe multi-organ disorders characterized by early childhood onset and developmental delay. Using patient blood samples and fibroblasts derived from a skin biopsy, we investigated the pathogenicity of the variant of unknown significance c.3G>T (p.1Met?) in the COQ7 gene and the effect of coenzyme Q10 supplementation in vitro. We showed that this variation leads to a severe decrease in COQ7 protein levels in the patient's fibroblasts, resulting in a decrease in coenzyme Q10 production and in the accumulation of 6-demethoxycoenzyme Q10, the COQ7 substrate. Interestingly, such accumulation was also found in the patient's plasma. Normal coenzyme Q10 and 6-demethoxycoenzyme Q10 levels were restored in vitro by using the coenzyme Q10 precursor 2,4-dihydroxybenzoic acid, thus bypassing the COQ7 requirement. Coenzyme Q10 biosynthesis deficiency is known to impair the mitochondrial respiratory chain. Seahorse experiments showed that the patient's cells mainly rely on glycolysis to maintain sufficient ATP production. Consistently, the replacement of glucose by galactose in the culture medium of these cells reduced their proliferation rate. Interestingly, normal proliferation was restored by coenzyme Q10 supplementation of the culture medium, suggesting a therapeutic avenue for these patients. Altogether, we have identified the first example of recessive distal hereditary motor neuropathy caused by a homozygous variation in the COQ7 gene, which should thus be included in the gene panels used to diagnose peripheral inherited neuropathies. Furthermore, 6-demethoxycoenzyme Q10 accumulation in the blood can be used to confirm the pathogenic nature of the mutation. Finally, supplementation with coenzyme Q10 or derivatives should be considered to prevent the progression of COQ7-related peripheral inherited neuropathy in diagnosed patients.
Keywords: COQ7; Coenzyme Q10; distal hereditary motor neuropathy.
© The Author(s) 2022. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
The authors report no competing interests.
Figures
Comment in
-
Axonal Charcot-Marie-Tooth disease due to COQ7 mutation: expanding the genetic and clinical spectrum.Brain. 2023 Dec 1;146(12):e117-e119. doi: 10.1093/brain/awad212. Brain. 2023. PMID: 37343138 No abstract available.
References
-
- Carroll AS, Burns J, Nicholson G, Kiernan MC, Vucic S. Inherited neuropathies. Semin Neurol. 2019;39:620–639. - PubMed
-
- Neuromuscular Home Page. Accessed 21 June 2022. https://neuromuscular.wustl.edu/
-
- Frasquet M, Rojas-García R, Argente-Escrig H, et al. Distal hereditary motor neuropathies: Mutation spectrum and genotype–phenotype correlation. Eur J Neurol. 2021;28:1334–1343. - PubMed
-
- Acosta MJ, Vazquez Fonseca L, Desbats MA, et al. Coenzyme Q biosynthesis in health and disease. Biochim Biophys Acta. 2016;185:1079–1085. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
