

The diagnosis of severe combined immunodeficiency: Implementation of the PIDTC 2022 Definitions
- PMID: 36456360
- PMCID: PMC9905305
- DOI: 10.1016/j.jaci.2022.10.021
The diagnosis of severe combined immunodeficiency: Implementation of the PIDTC 2022 Definitions
Abstract
Background: Shearer et al in 2014 articulated well-defined criteria for the diagnosis and classification of severe combined immunodeficiency (SCID) as part of the Primary Immune Deficiency Treatment Consortium's (PIDTC's) prospective and retrospective studies of SCID.
Objective: Because of the advent of newborn screening for SCID and expanded availability of genetic sequencing, revision of the PIDTC 2014 Criteria was needed.
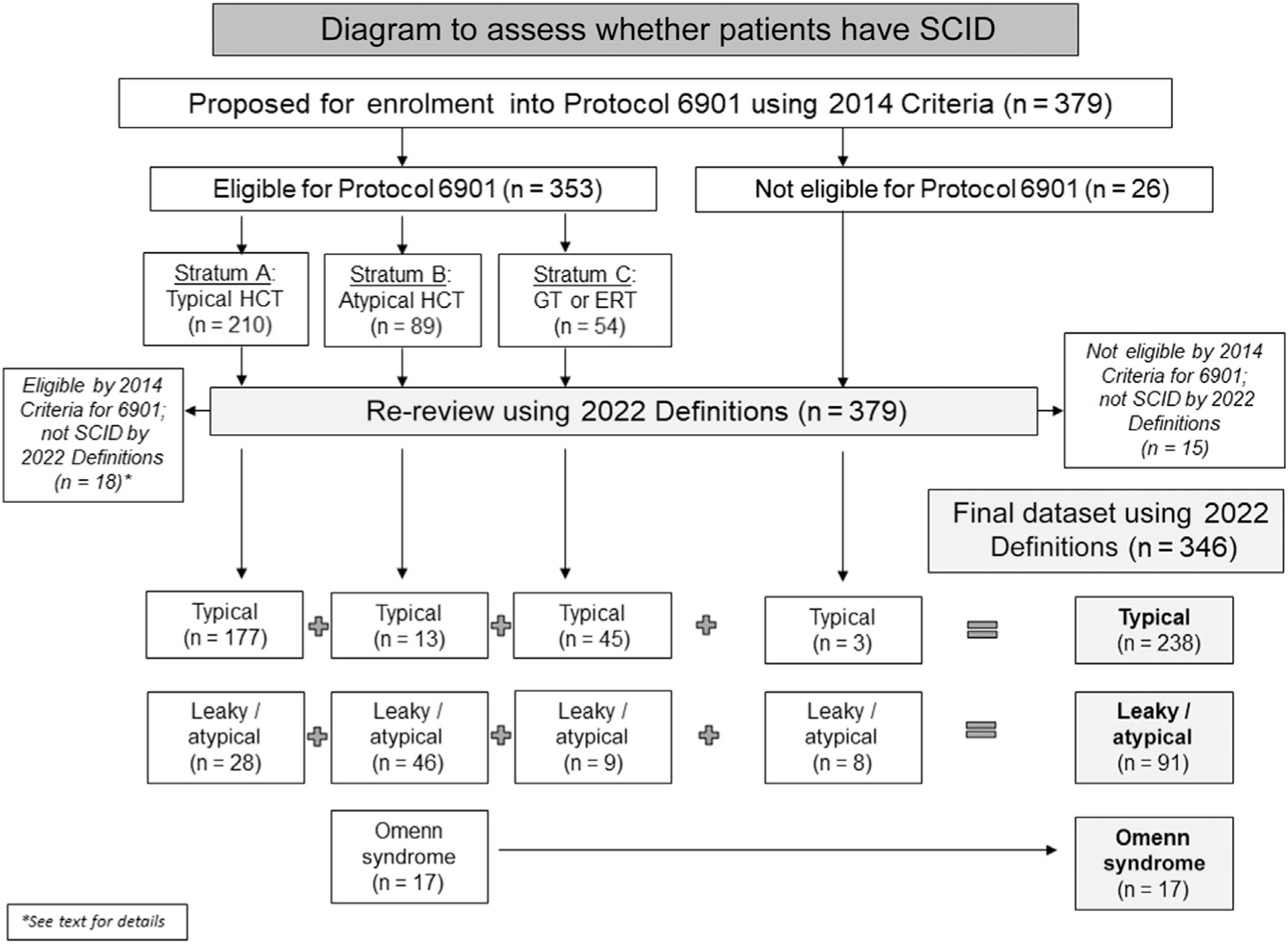
Methods: We developed and tested updated PIDTC 2022 SCID Definitions by analyzing 379 patients proposed for prospective enrollment into Protocol 6901, focusing on the ability to distinguish patients with various SCID subtypes.
Results: According to PIDTC 2022 Definitions, 18 of 353 patients eligible per 2014 Criteria were considered not to have SCID, whereas 11 of 26 patients ineligible per 2014 Criteria were determined to have SCID. Of note, very low numbers of autologous T cells (<0.05 × 109/L) characterized typical SCID under the 2022 Definitions. Pathogenic variant(s) in SCID-associated genes was identified in 93% of patients, with 7 genes (IL2RG, RAG1, ADA, IL7R, DCLRE1C, JAK3, and RAG2) accounting for 89% of typical SCID. Three genotypes (RAG1, ADA, and RMRP) accounted for 57% of cases of leaky/atypical SCID; there were 13 other rare genotypes. Patients with leaky/atypical SCID were more likely to be diagnosed at more than age 1 year than those with typical SCID lacking maternal T cells: 20% versus 1% (P < .001). Although repeat testing proved important, an initial CD3 T-cell count of less than 0.05 × 109/L differentiated cases of typical SCID lacking maternal cells from leaky/atypical SCID: 97% versus 7% (P < .001).
Conclusions: The PIDTC 2022 Definitions describe SCID and its subtypes more precisely than before, facilitating analyses of SCID characteristics and outcomes.
Keywords: Omenn syndrome; SCID; Severe combined immunodeficiency; leaky/atypical SCID; newborn screening; typical SCID.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Figures


References
-
- Dvorak CC, Cowan MJ, Logan BR, Notarangelo LD, Griffith LM, Puck JM, et al. The natural history of children with severe combined immunodeficiency: baseline features of the first fifty patients of the Primary Immune Deficiency Treatment Consortium prospective study 6901. J Clin Immunol 2013;33:1156–64. - PMC - PubMed
-
- Shearer WT, Dunn E, Notarangelo LD, Dvorak CC, Puck JM, Logan BR, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. J Allergy Clin Immunol 2014;133:1092–8. - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- U01 TR001263/TR/NCATS NIH HHS/United States
- U24 CA076518/CA/NCI NIH HHS/United States
- U54 AI082973/AI/NIAID NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- P30 CA042014/CA/NCI NIH HHS/United States
- U01 HL069294/HL/NHLBI NIH HHS/United States
- U54 NS064808/NS/NINDS NIH HHS/United States
- ZIA AI001222/ImNIH/Intramural NIH HHS/United States
- UG1 HL069254/HL/NHLBI NIH HHS/United States
- U10 HL069254/HL/NHLBI NIH HHS/United States
- R13 AI094943/AI/NIAID NIH HHS/United States
- U01 AI126612/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Research Materials
