

Abundant copathologies of polyglucosan bodies, frontotemporal lobar degeneration with TDP-43 inclusions and ageing-related tau astrogliopathy in a family with a GBE1 mutation
- PMID: 36456471
- PMCID: PMC9992093
- DOI: 10.1111/nan.12865
Abundant copathologies of polyglucosan bodies, frontotemporal lobar degeneration with TDP-43 inclusions and ageing-related tau astrogliopathy in a family with a GBE1 mutation
Abstract
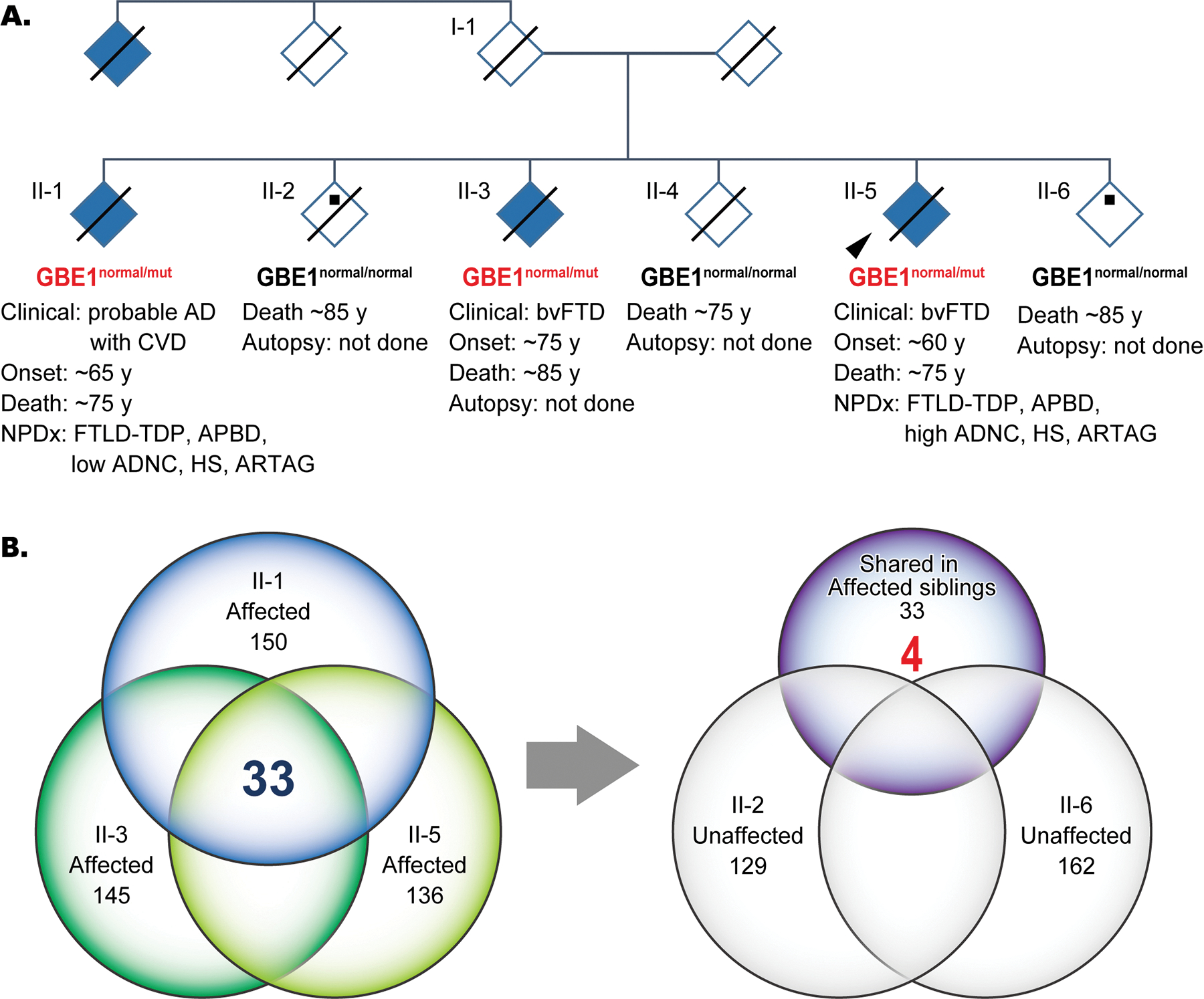
Aims: Adult polyglucosan body disease (APBD) is a progressive neurogenetic disorder caused by 1,4-alpha-glucan branching enzyme 1 (GBE1) mutation with an accumulation of polyglucosan bodies (PBs) in the central and peripheral nervous systems as a pathological hallmark. Here, we report two siblings in a family with a GBE1 mutation with prominent frontotemporal lobar degeneration with TAR DNA-binding protein 43 (FTLD-TDP) and ageing-related tau astrogliopathy (ARTAG) copathologies with PBs in the central nervous system.
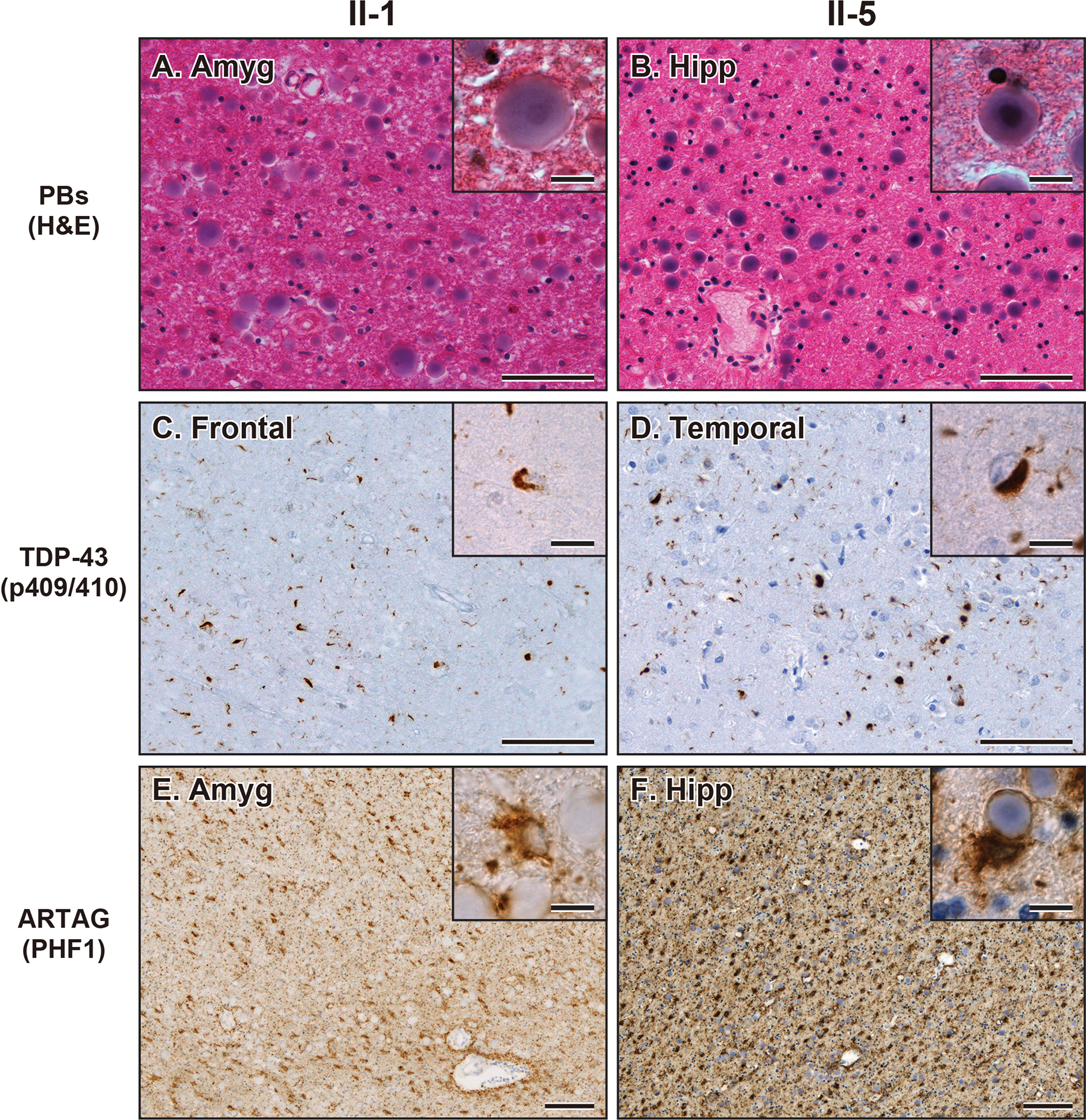
Methods: Whole-genome sequencing (WGS) followed by Sanger sequencing (SS) was performed on three affected and two unaffected siblings in a pedigree diagnosed with familial frontotemporal dementia. Out of the affected siblings, autopsies were conducted on two cases, and brain samples were used for biochemical and histological analyses. Brain sections were stained with haematoxylin and eosin and immunostained with antibodies against ubiquitin, tau, amyloid β, α-synuclein, TDP-43 and fused in sarcoma (FUS).
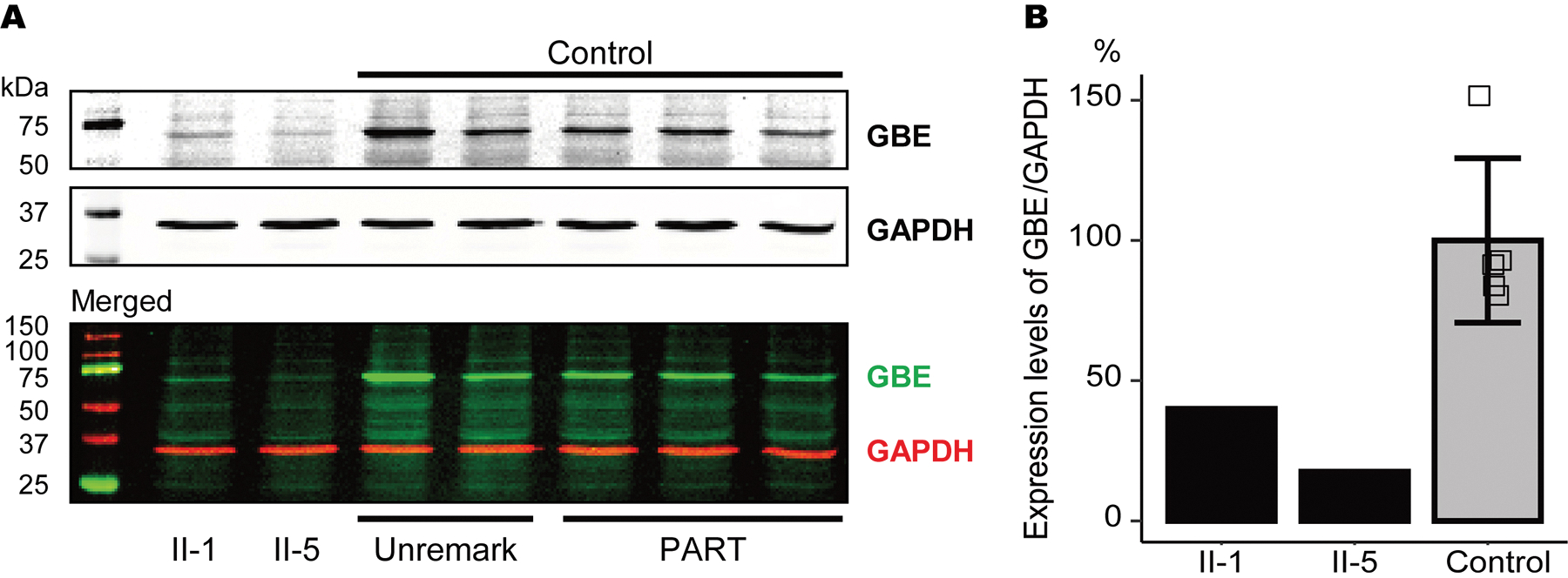
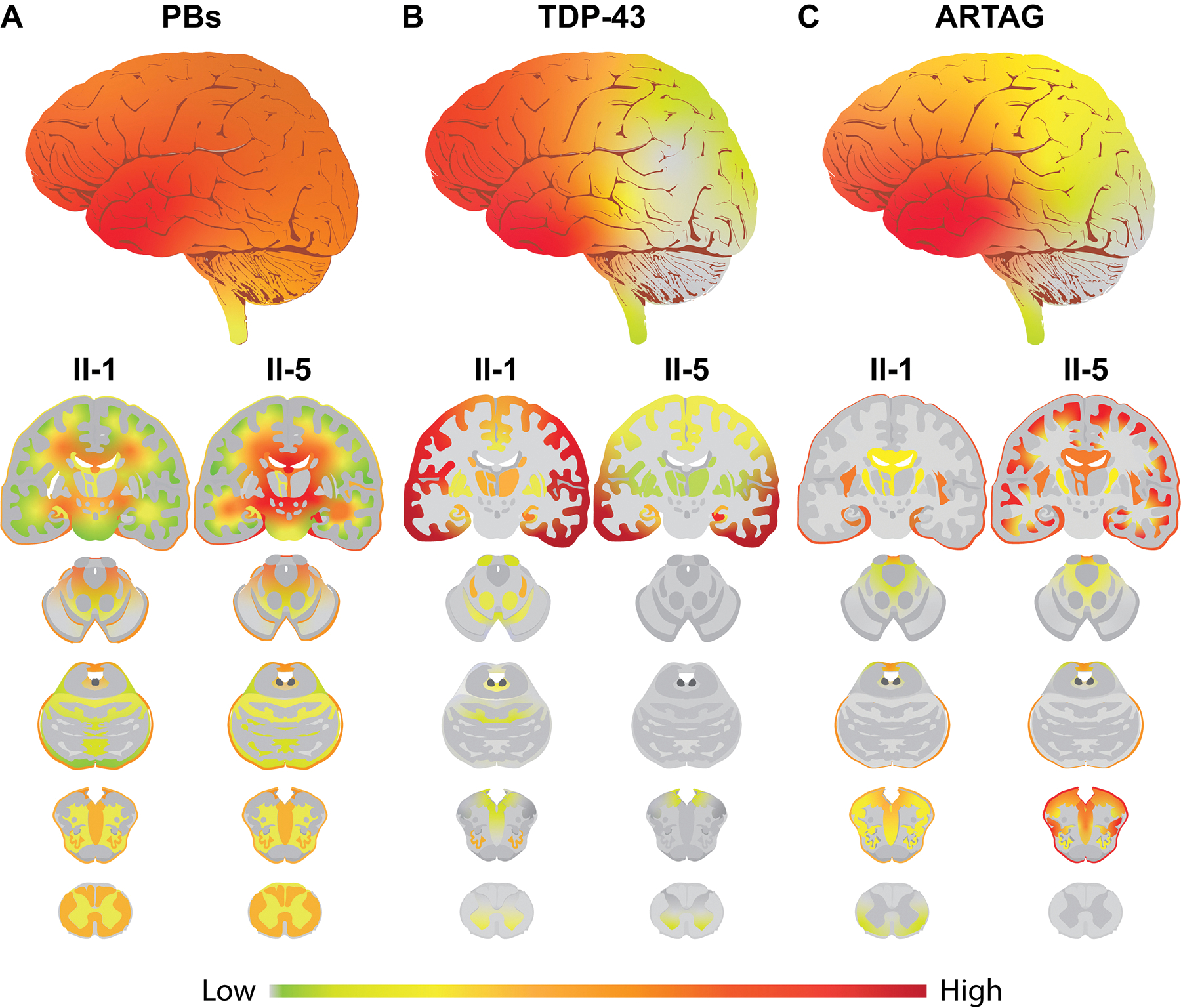
Results: A novel single nucleotide deletion in GBE1, c.1280delG, was identified, which is predicted to result in a reading frameshift, p.Gly427Glufs*9. This variant segregated with disease in the family, is absent from population databases and is predicted to cause loss of function, a known genetic mechanism for APBD. The affected siblings showed a greater than 50% decrease in GBE protein levels. Immunohistochemical analysis revealed widespread FTLD-TDP (type A) and ARTAG pathologies as well as PBs in the brains of two affected siblings for whom an autopsy was performed.
Conclusions: This is the first report of a family with several individuals with a FTD clinical phenotype and underlying copathologies of APBD, FTLD-TDP and ARTAG with a segregating GBE1 loss-of-function mutation in affected siblings. The finding of copathologies of APBD and FTLD-TDP suggests these processes may share a disease mechanism resulting from this GBE1 mutation.
Keywords: ARTAG; FTLD-TDP; GBE1; adult polyglucosan body disease; corpora amylacea.
© 2022 British Neuropathological Society.
Figures
References
-
- Mochel F, Schiffmann R, Steenweg ME, Akman HO, Wallace M, Sedel F, Laforêt P, Levy R, Powers MJ, Demeret S, Maisonobe T, Froissart R, Nobrega B, Fogel BL, Natowicz MR, Lubetzki C, Durr A, Brice A, Rosenmann H, Barash V, Kakhlon O, Gomori MJ, Knaap MSvd, Lossos A. Adult polyglucosan body disease: Natural History and Key Magnetic Resonance Imaging Findings. Annals of neurology 2012; 72: 433–41 - PMC - PubMed
-
- Akman HO, Kakhlon O, Coku J, Peverelli L, Rosenmann H, Rozenstein-Tsalkovich L, Turnbull J, Meiner V, Chama L, Lerer I, Shpitzen S, Leitersdorf E, Paradas C, Wallace M, Schiffmann R, DiMauro S, Lossos A, Minassian BA. Deep Intronic GBE1 Mutation in Manifesting Heterozygous Patients With Adult Polyglucosan Body Disease. Jama Neurol 2015; 72: 441–5 - PubMed
-
- Lossos A, Meiner Z, Barash V, Soffer D, Schlesinger I, Abramsky O, Argov Z, Shpitzen S, Meiner V. Adult polyglucosan body disease in Ashkenazi Jewish patients carrying the Tyr329Ser mutation in the glycogen-branching enzyme gene. Annals of neurology 1998; 44: 867–72 - PubMed
-
- Akman OH, Kurt Y, Wallace M, Schiffmann R, DiMauro S. Compound heterozygosis of a splice site and the common Ashkenazi Jewish mutation in GBE1 causes adult onset polyglucosan body disease. (P2.026). Neurology 2015; 84: P 2.026
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
