

Genomic analysis of sewage from 101 countries reveals global landscape of antimicrobial resistance
- PMID: 36456547
- PMCID: PMC9715550
- DOI: 10.1038/s41467-022-34312-7
Genomic analysis of sewage from 101 countries reveals global landscape of antimicrobial resistance
Erratum in
-
Author Correction: Genomic analysis of sewage from 101 countries reveals global landscape of antimicrobial resistance.Nat Commun. 2023 Jan 12;14(1):178. doi: 10.1038/s41467-023-35890-w. Nat Commun. 2023. PMID: 36635285 Free PMC article. No abstract available.
Abstract
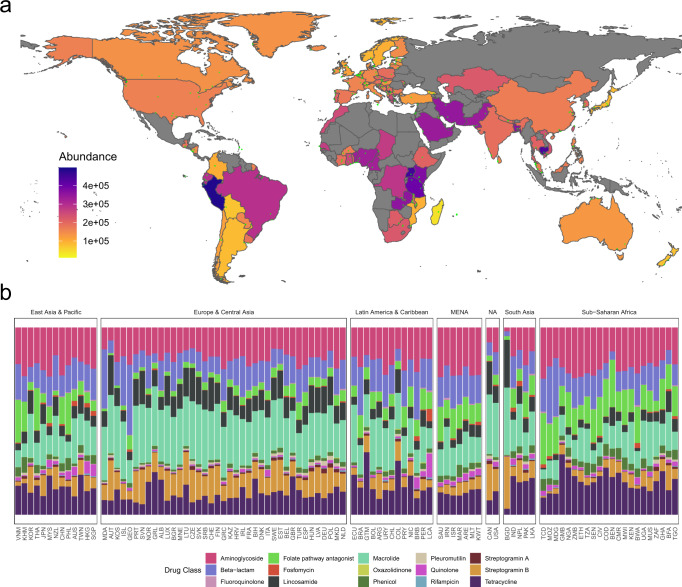
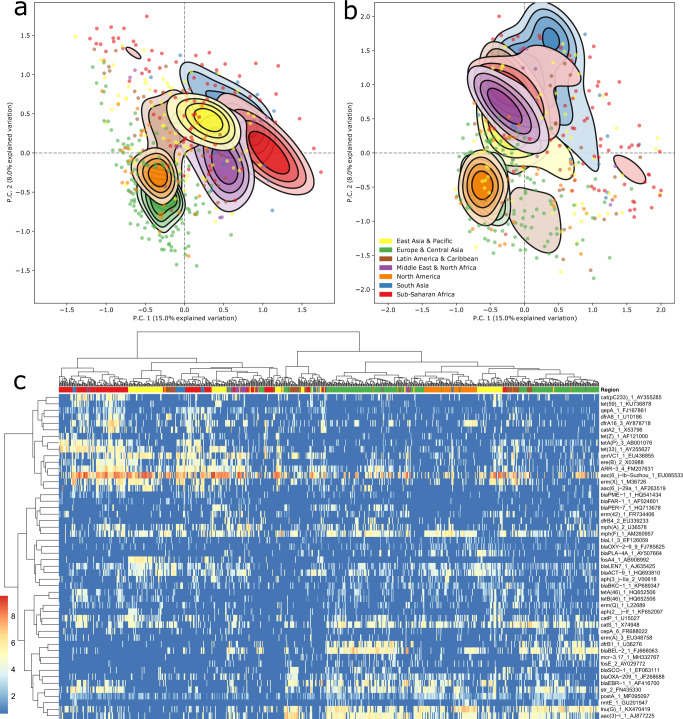
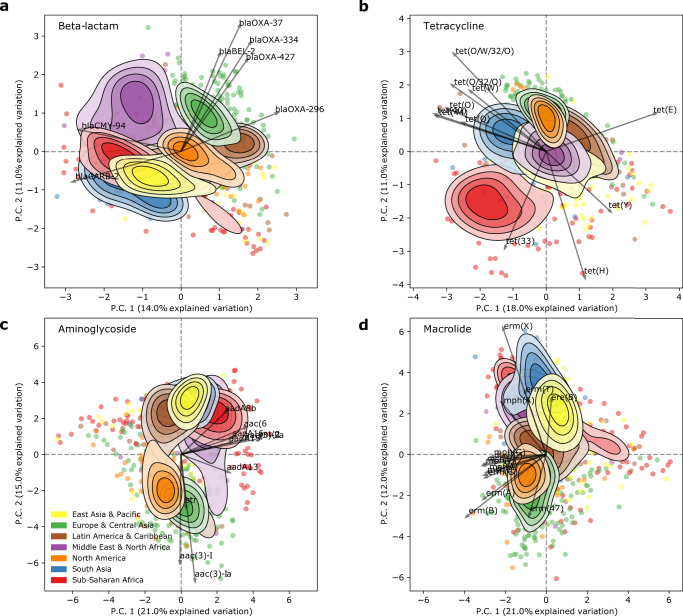
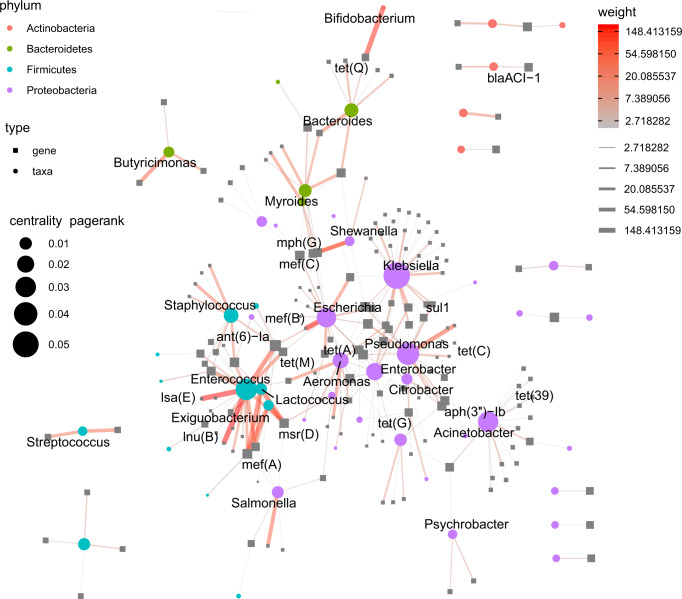
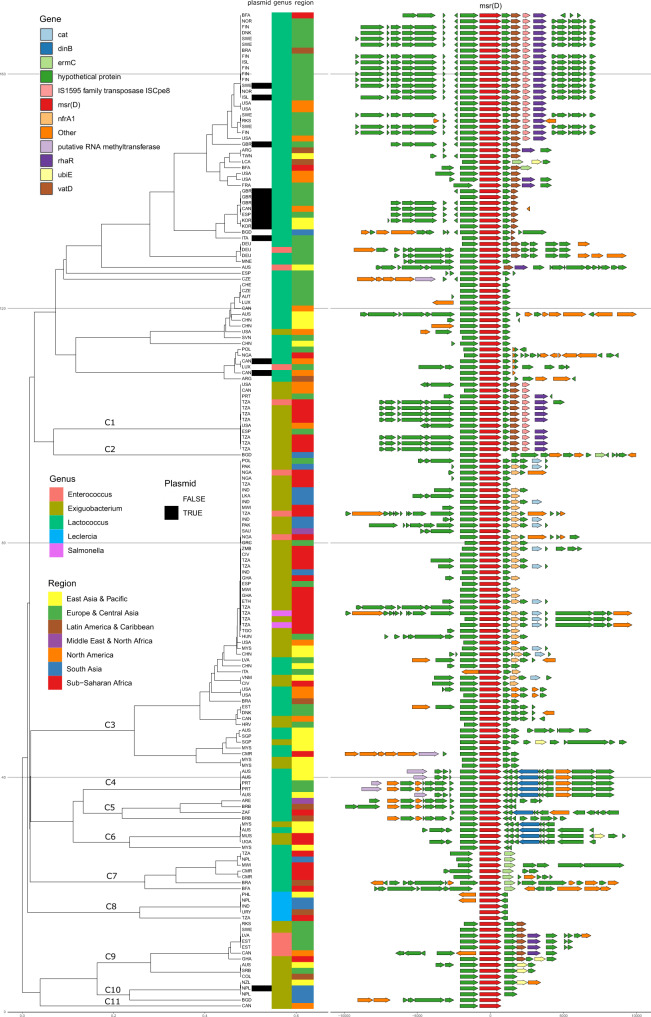
Antimicrobial resistance (AMR) is a major threat to global health. Understanding the emergence, evolution, and transmission of individual antibiotic resistance genes (ARGs) is essential to develop sustainable strategies combatting this threat. Here, we use metagenomic sequencing to analyse ARGs in 757 sewage samples from 243 cities in 101 countries, collected from 2016 to 2019. We find regional patterns in resistomes, and these differ between subsets corresponding to drug classes and are partly driven by taxonomic variation. The genetic environments of 49 common ARGs are highly diverse, with most common ARGs carried by multiple distinct genomic contexts globally and sometimes on plasmids. Analysis of flanking sequence revealed ARG-specific patterns of dispersal limitation and global transmission. Our data furthermore suggest certain geographies are more prone to transmission events and should receive additional attention.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- VEO. VEO report on mutations and variation in publicly shared SARS- CoV-2 raw sequencing data. https://www.veo-europe.eu/-/media/Sites/veo-europe/Library/VEO-COVID-19-... 874735, (2021).
-
- Volz, E. et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature593, 266–269 (2021). - PubMed
-
- O’Neill, J. Tackling drug-resistant infections globally: final report and recommendations. Rev. Antimicrob. Resist. 84 (2016) 10.1016/j.jpha.2015.11.005.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
