

A unified computational framework for single-cell data integration with optimal transport
- PMID: 36456571
- PMCID: PMC9715710
- DOI: 10.1038/s41467-022-35094-8
A unified computational framework for single-cell data integration with optimal transport
Abstract
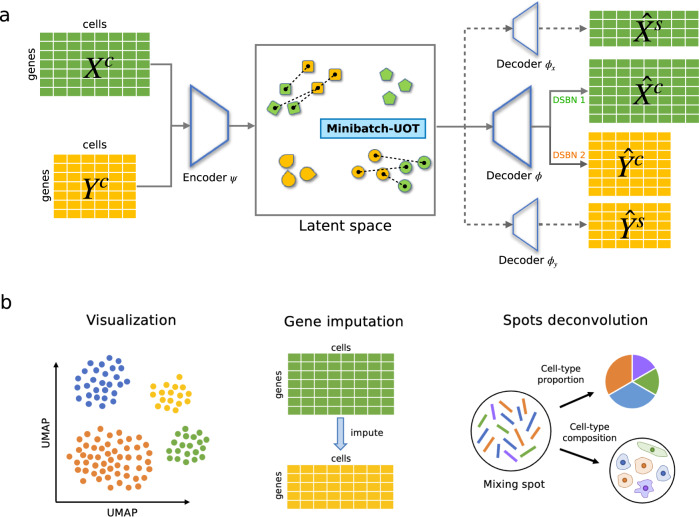
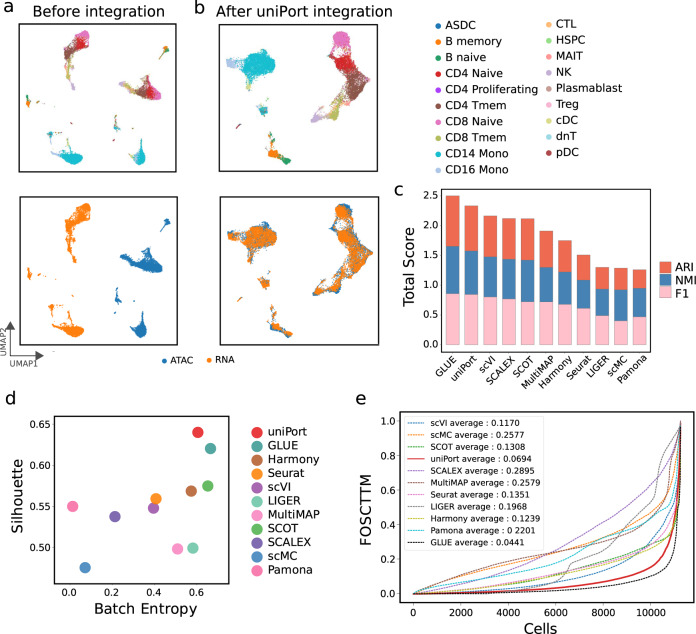
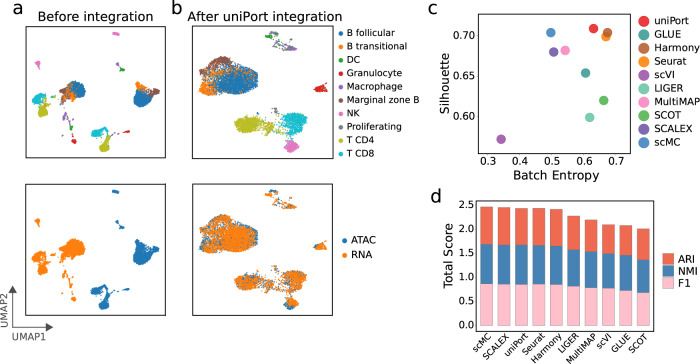
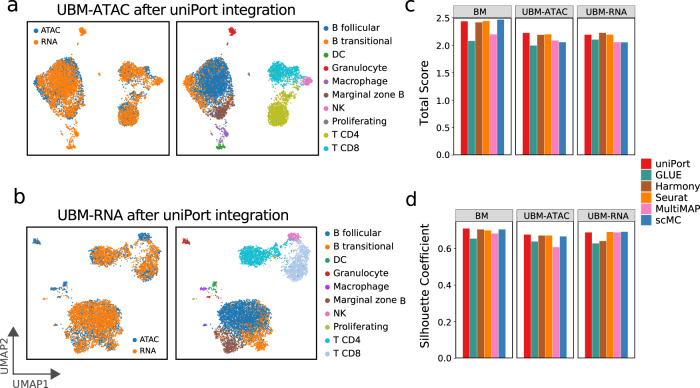
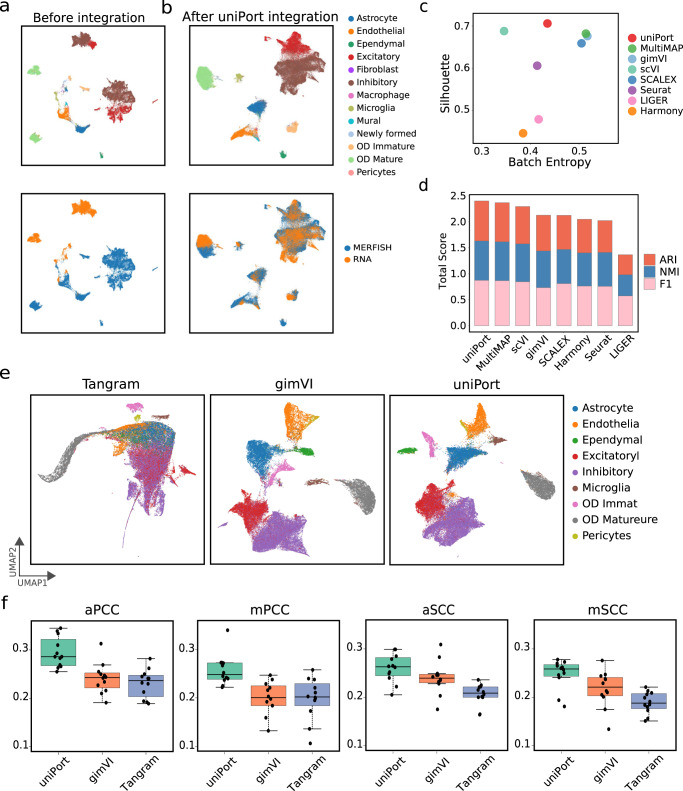
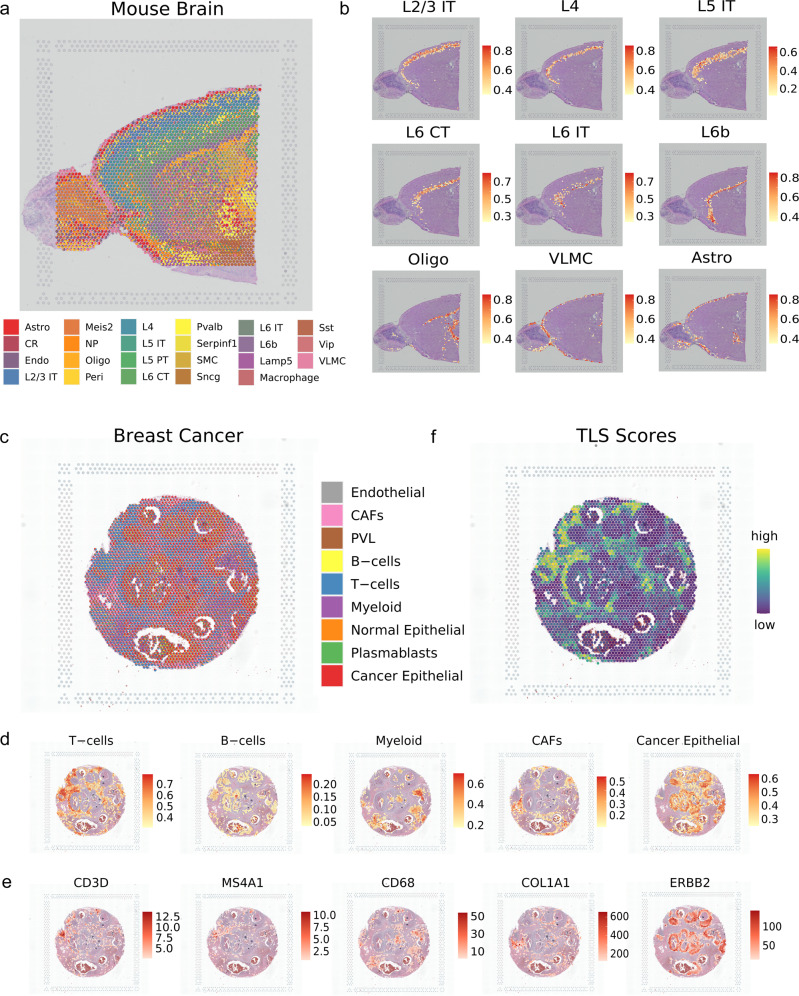
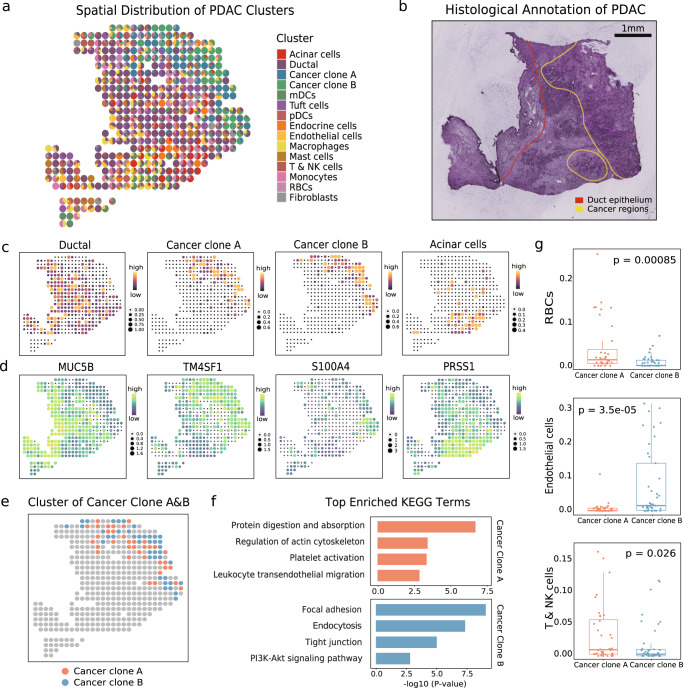
Single-cell data integration can provide a comprehensive molecular view of cells. However, how to integrate heterogeneous single-cell multi-omics as well as spatially resolved transcriptomic data remains a major challenge. Here we introduce uniPort, a unified single-cell data integration framework that combines a coupled variational autoencoder (coupled-VAE) and minibatch unbalanced optimal transport (Minibatch-UOT). It leverages both highly variable common and dataset-specific genes for integration to handle the heterogeneity across datasets, and it is scalable to large-scale datasets. uniPort jointly embeds heterogeneous single-cell multi-omics datasets into a shared latent space. It can further construct a reference atlas for gene imputation across datasets. Meanwhile, uniPort provides a flexible label transfer framework to deconvolute heterogeneous spatial transcriptomic data using an optimal transport plan, instead of embedding latent space. We demonstrate the capability of uniPort by applying it to integrate a variety of datasets, including single-cell transcriptomics, chromatin accessibility, and spatially resolved transcriptomic data.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Chen X, et al. Cell type annotation of single-cell chromatin accessibility data via supervised bayesian embedding. Nat. Mach. Intelligence. 2022;4:116–126. doi: 10.1038/s42256-021-00432-w. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
