

The genomic and immune landscape of long-term survivors of high-grade serous ovarian cancer
- PMID: 36456881
- PMCID: PMC10478425
- DOI: 10.1038/s41588-022-01230-9
The genomic and immune landscape of long-term survivors of high-grade serous ovarian cancer
Abstract
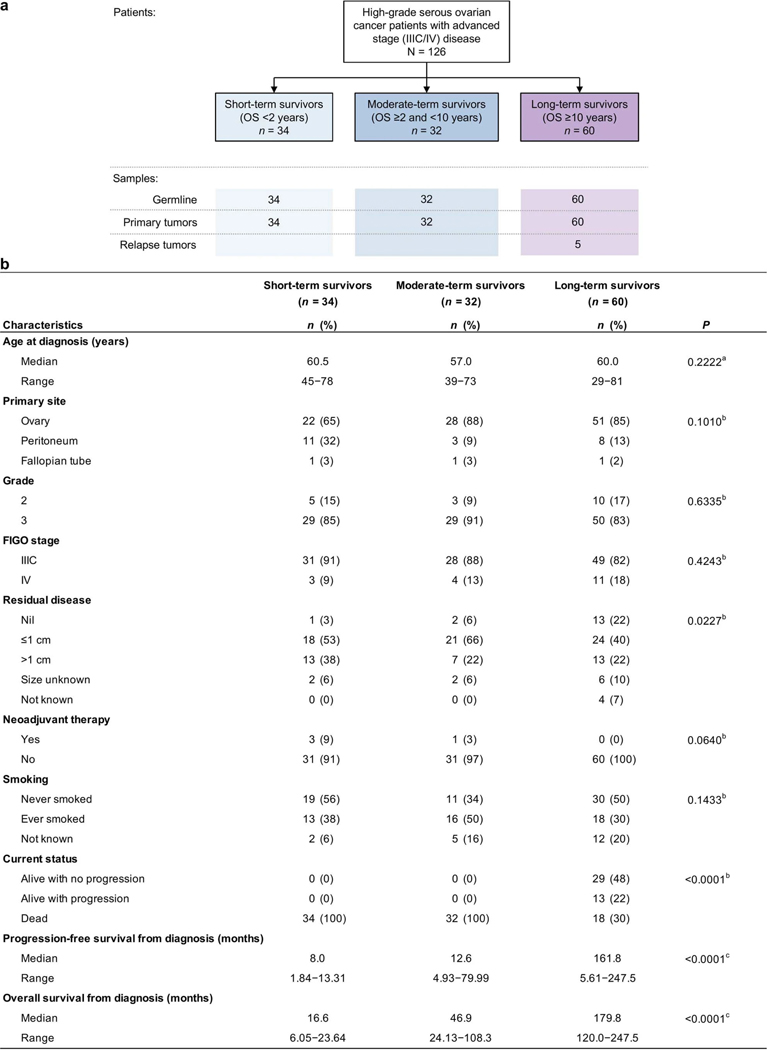
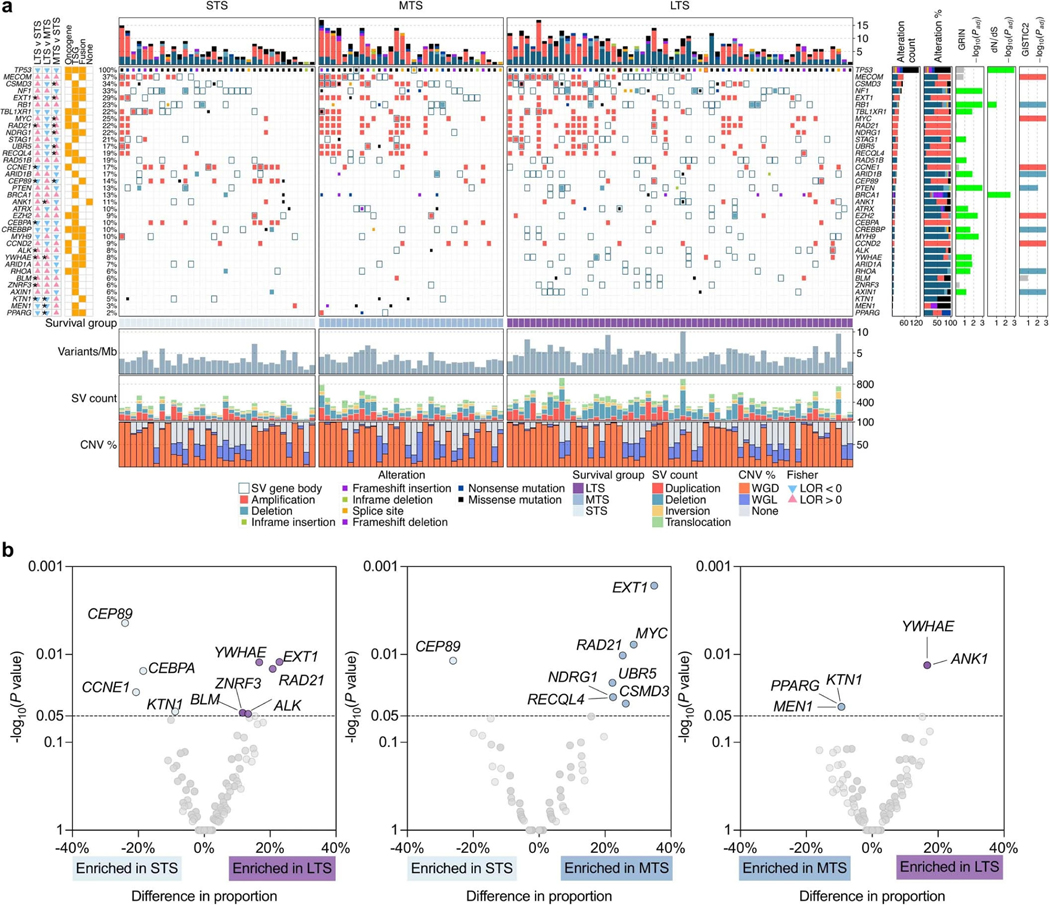
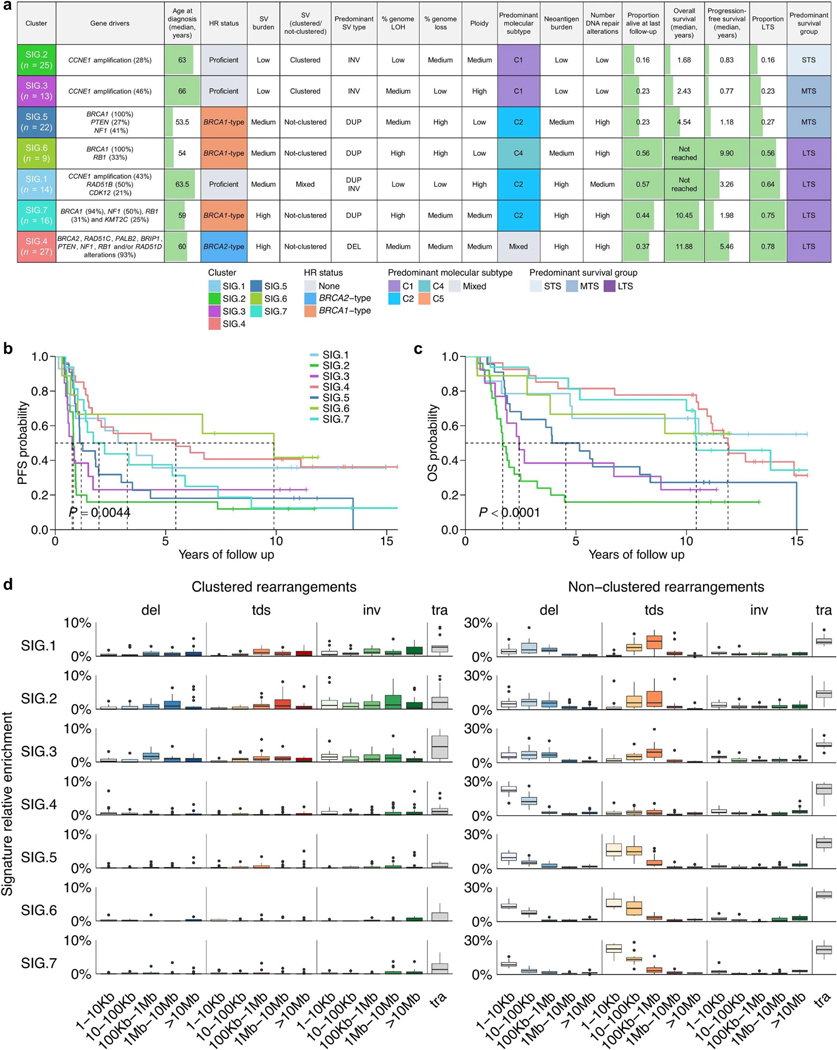
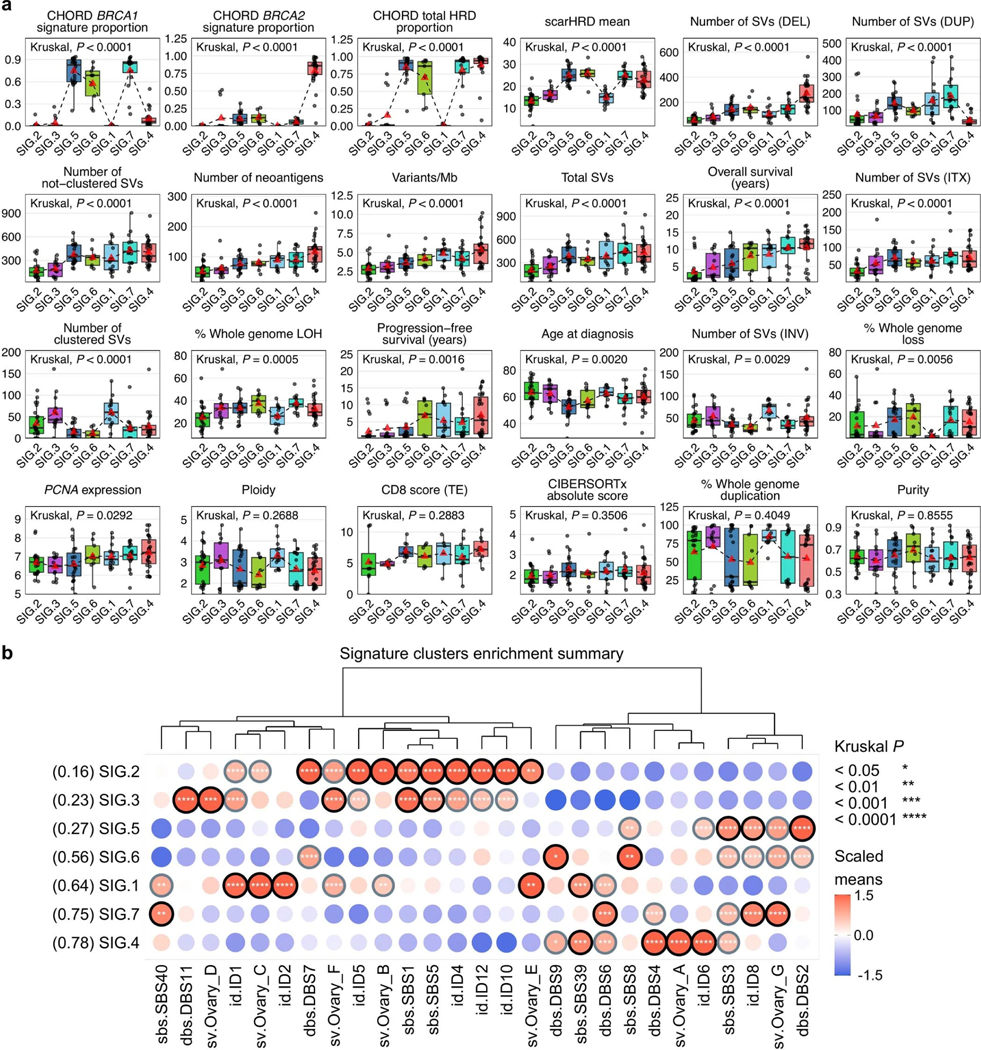
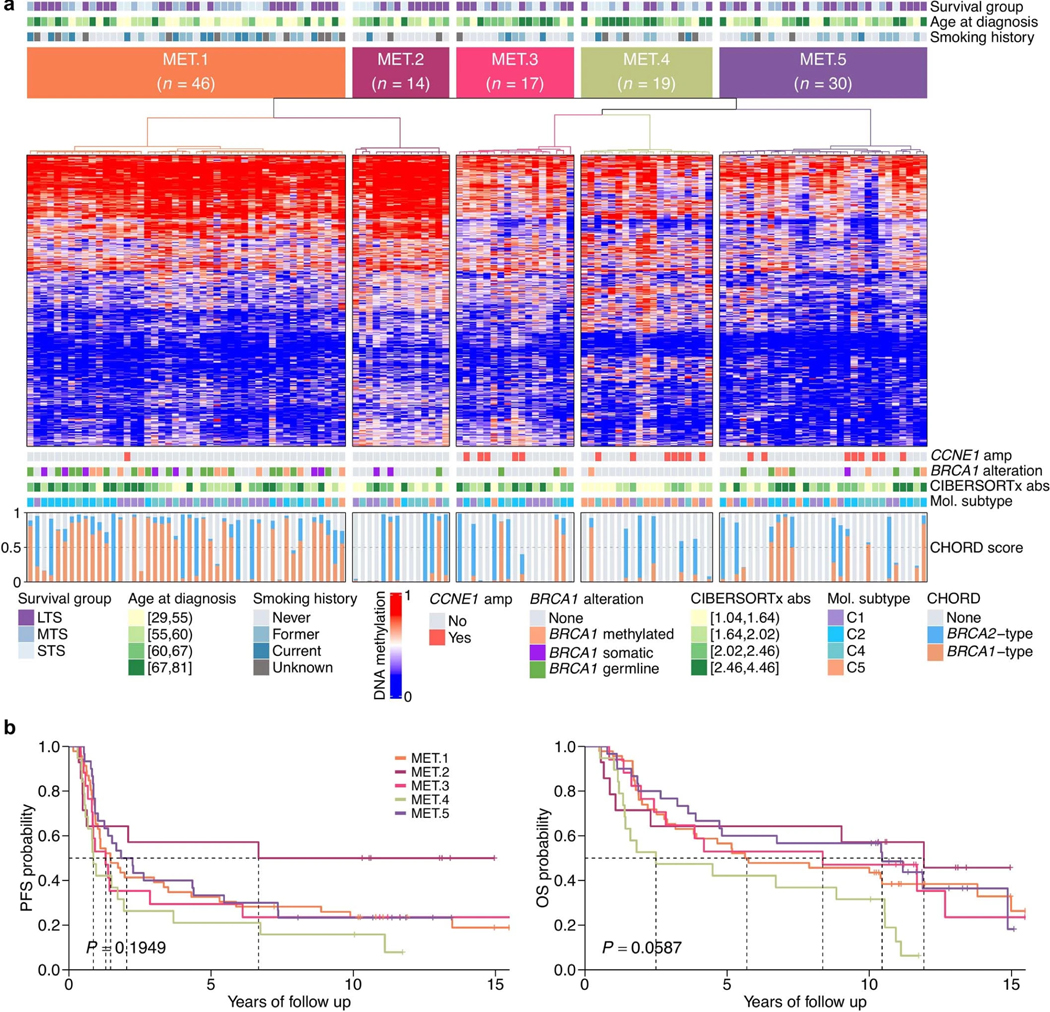
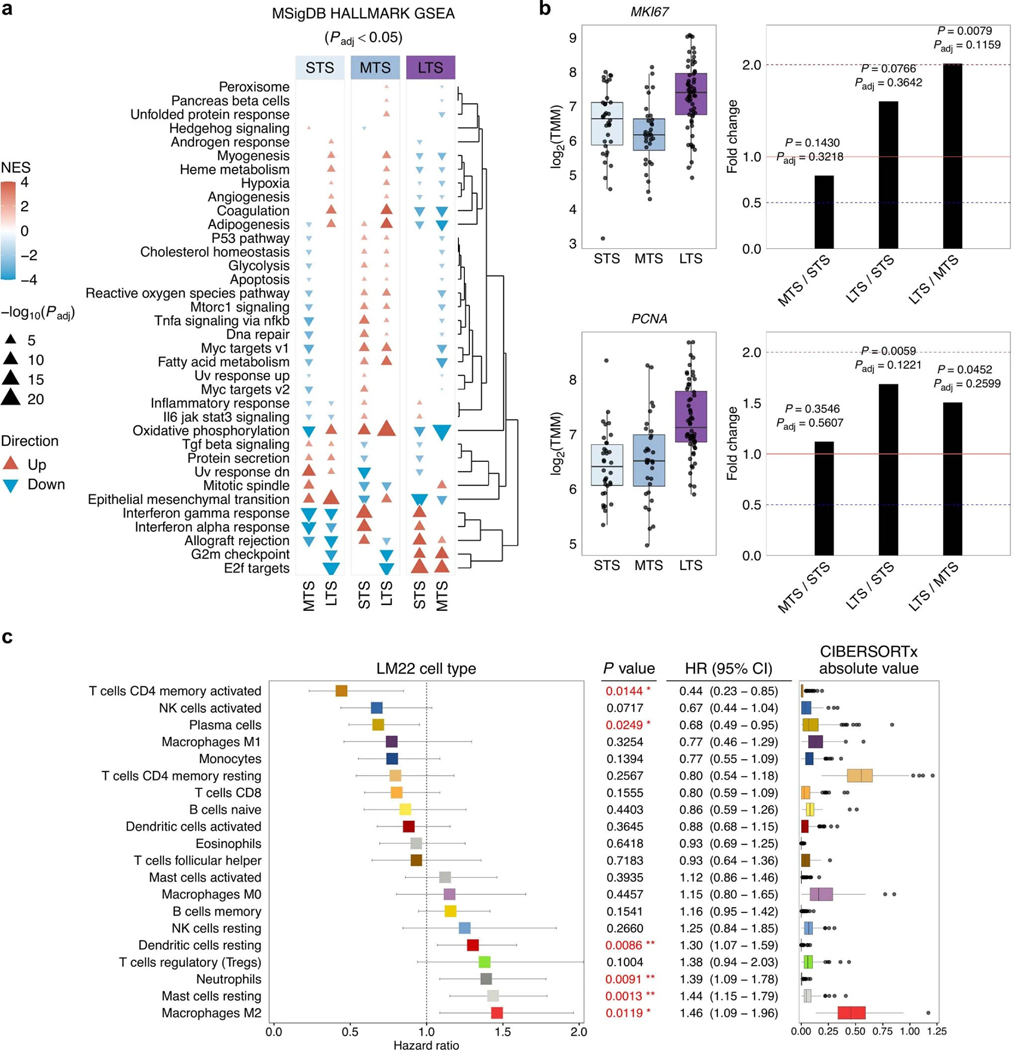
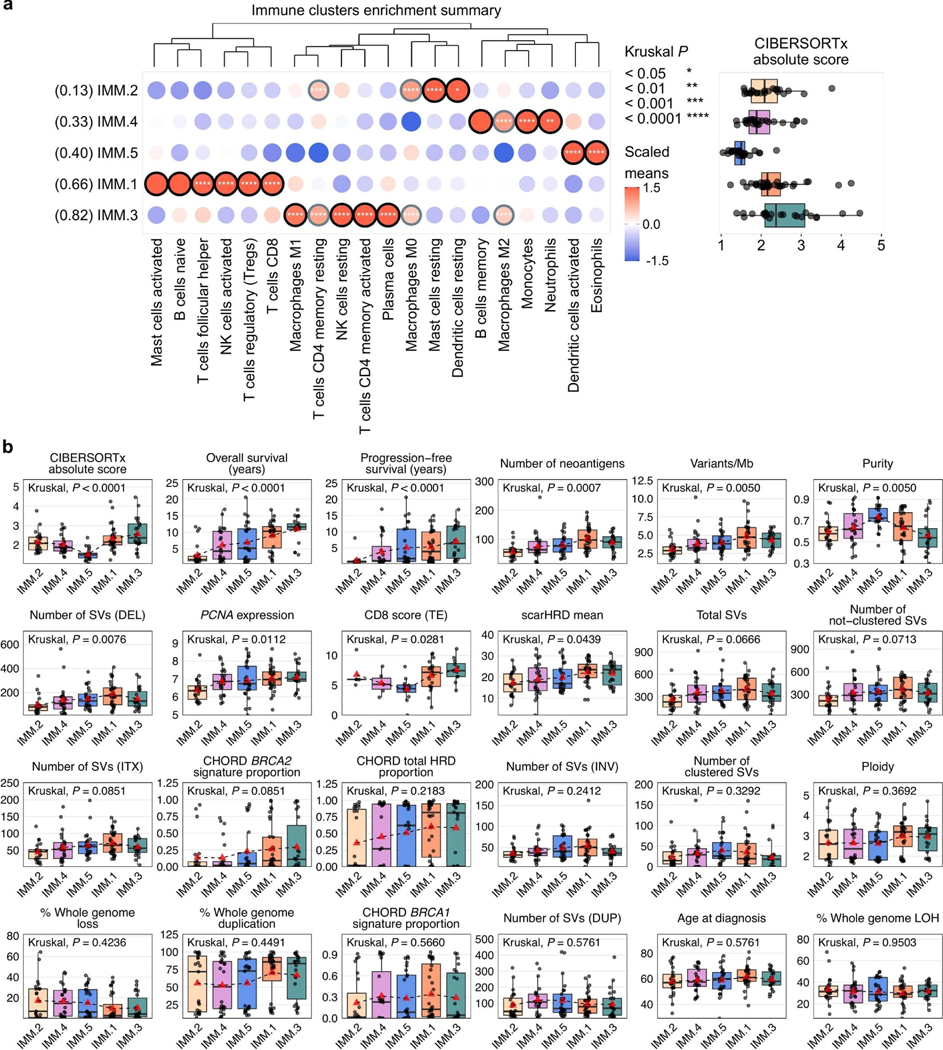
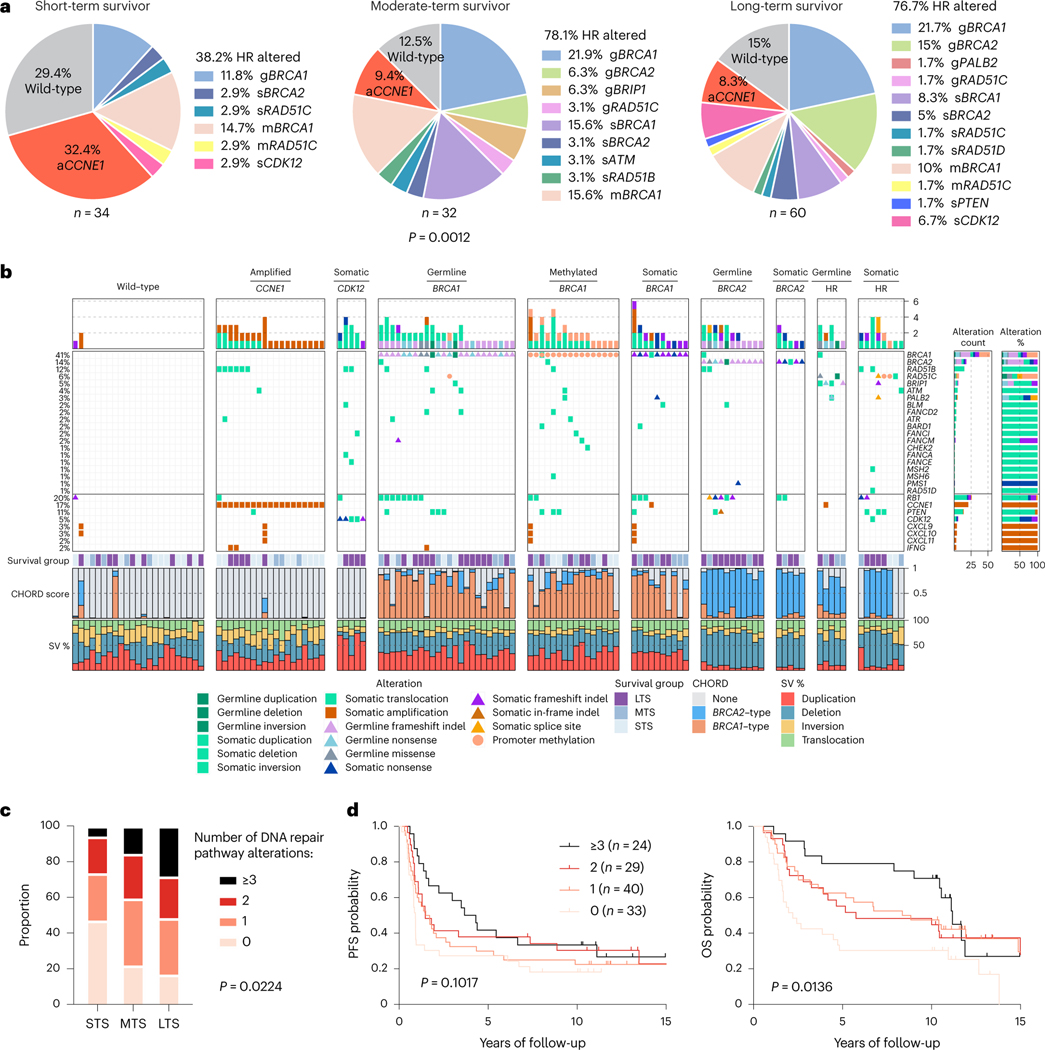
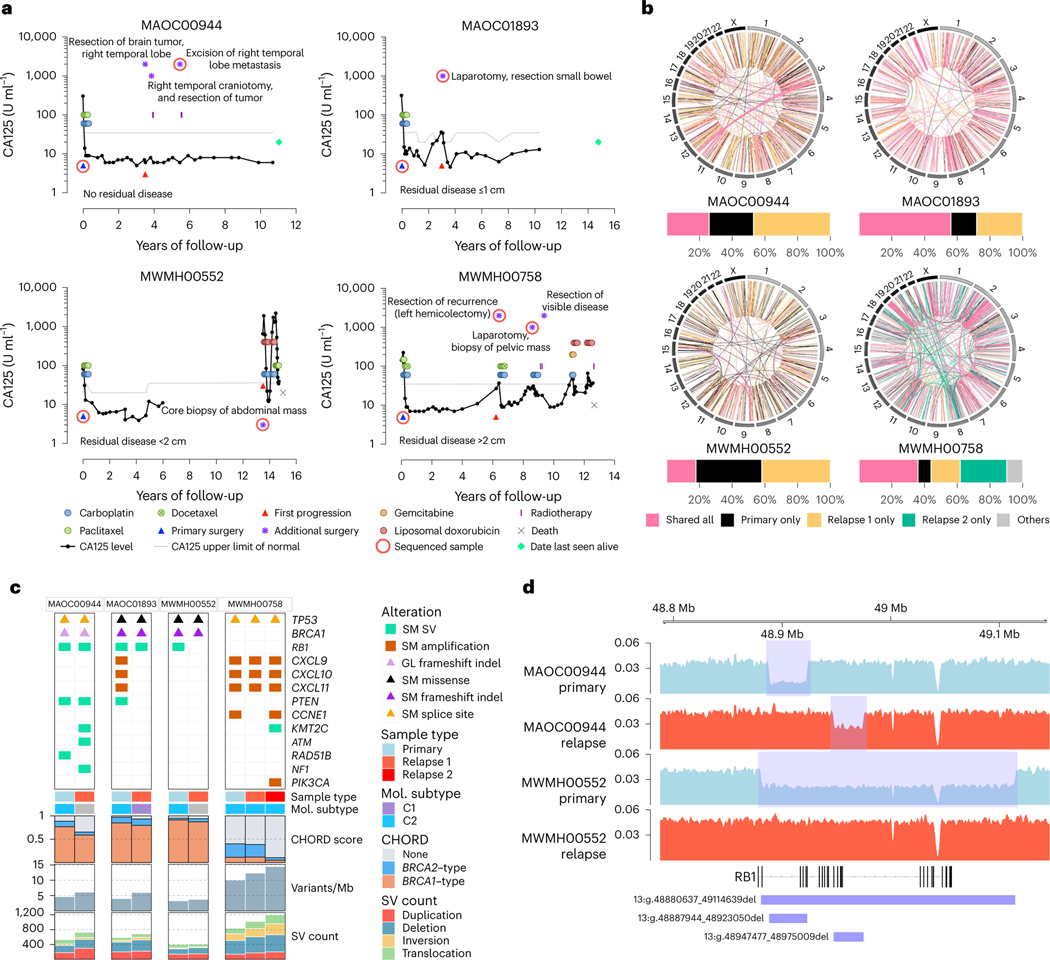
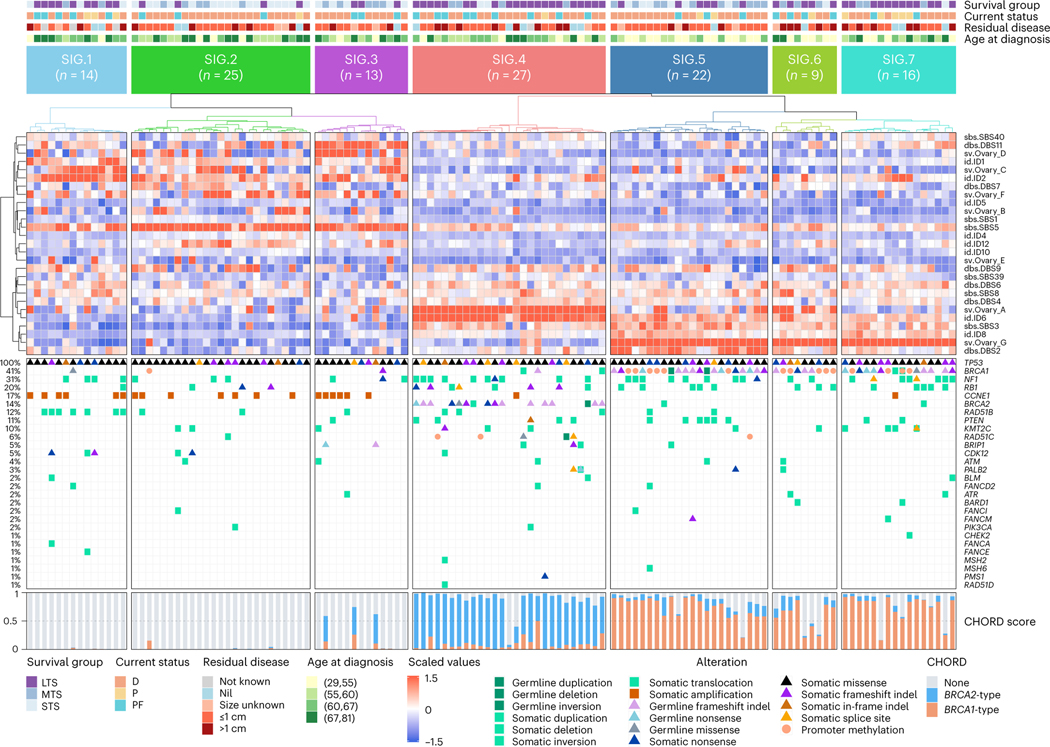
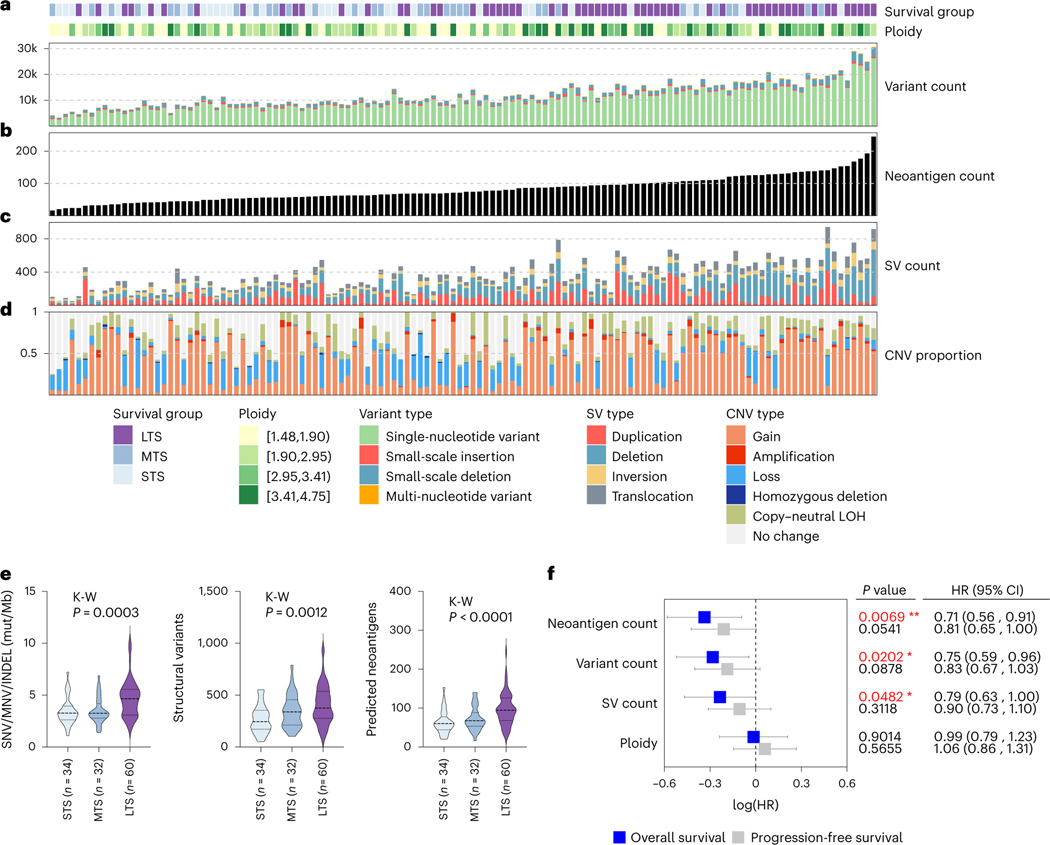
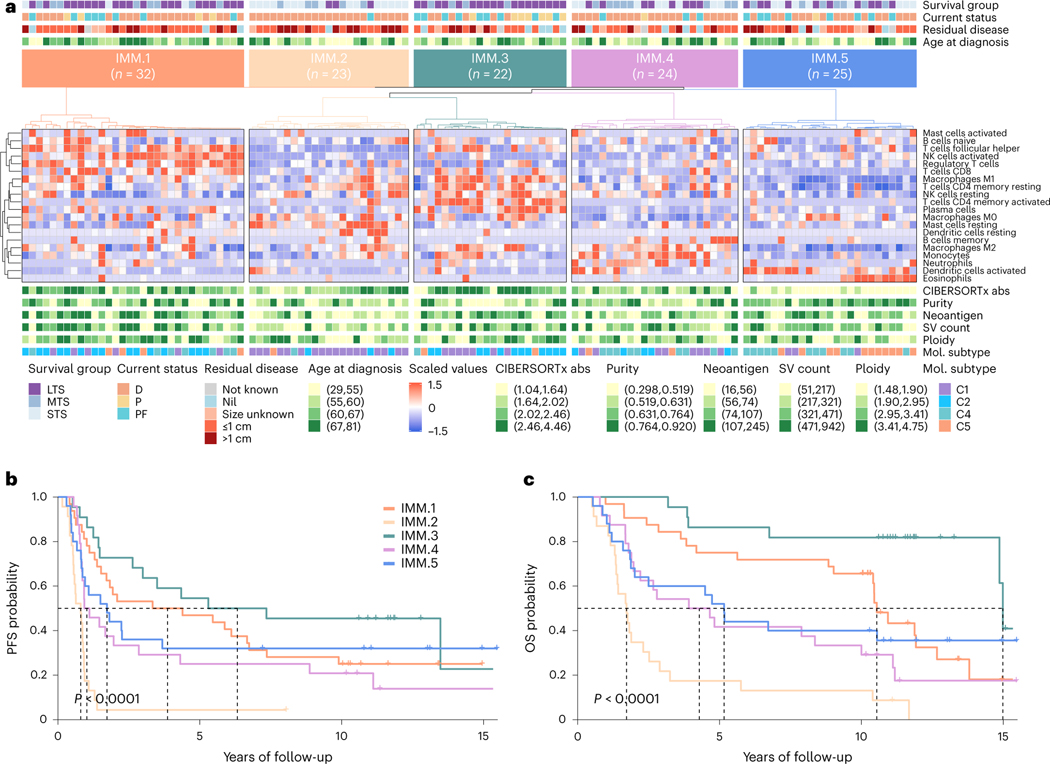
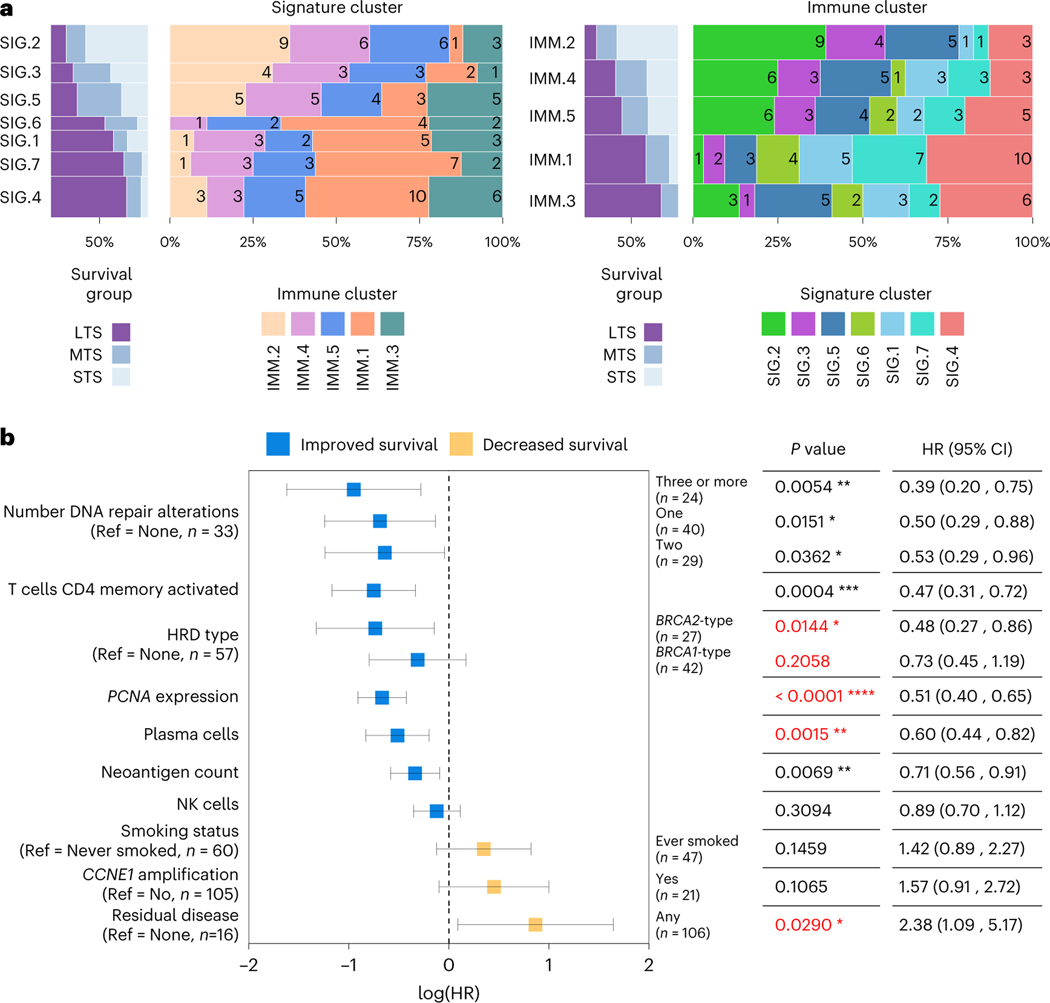
Fewer than half of all patients with advanced-stage high-grade serous ovarian cancers (HGSCs) survive more than five years after diagnosis, but those who have an exceptionally long survival could provide insights into tumor biology and therapeutic approaches. We analyzed 60 patients with advanced-stage HGSC who survived more than 10 years after diagnosis using whole-genome sequencing, transcriptome and methylome profiling of their primary tumor samples, comparing this data to 66 short- or moderate-term survivors. Tumors of long-term survivors were more likely to have multiple alterations in genes associated with DNA repair and more frequent somatic variants resulting in an increased predicted neoantigen load. Patients clustered into survival groups based on genomic and immune cell signatures, including three subsets of patients with BRCA1 alterations with distinctly different outcomes. Specific combinations of germline and somatic gene alterations, tumor cell phenotypes and differential immune responses appear to contribute to long-term survival in HGSC.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
The remaining authors declare no competing interests.
Figures


References
-
- Hoppenot C, Eckert MA, Tienda SM & Lengyel E. Who are the long-term survivors of high grade serous ovarian cancer? Gynecol. Oncol 148, 204–212 (2018). - PubMed
-
- Fago-Olsen CL et al. Does neoadjuvant chemotherapy impair long-term survival for ovarian cancer patients? A nationwide Danish study. Gynecol. Oncol 132, 292–298 (2014). - PubMed
-
- Chi DS et al. What is the optimal goal of primary cytoreductive surgery for bulky stage IIIC epithelial ovarian carcinoma (EOC)? Gynecol. Oncol 103, 559–564 (2006). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
