

Functional characterization of 5p15.33 risk locus in uveal melanoma reveals rs452384 as a functional variant and NKX2.4 as an allele-specific interactor
- PMID: 36459980
- PMCID: PMC9748249
- DOI: 10.1016/j.ajhg.2022.11.004
Functional characterization of 5p15.33 risk locus in uveal melanoma reveals rs452384 as a functional variant and NKX2.4 as an allele-specific interactor
Abstract
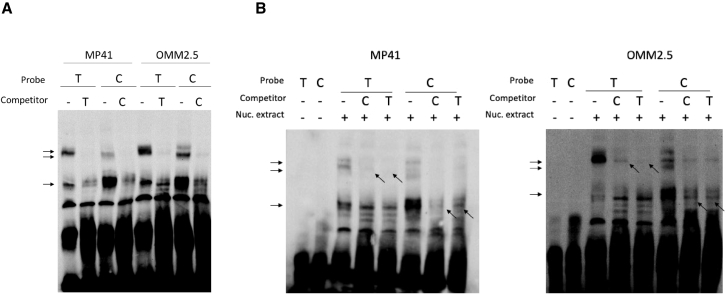
The TERT/CLPTM1L risk locus on chromosome 5p15.33 is a pleiotropic cancer risk locus in which multiple independent risk alleles have been identified, across well over ten cancer types. We previously conducted a genome-wide association study in uveal melanoma (UM), which uncovered a role for the TERT/CLPTM1L risk locus in this intraocular tumor and identified multiple highly correlated risk alleles. Aiming to unravel the biological mechanisms in UM of this locus, which contains a domain enriched in active chromatin marks and enhancer elements, we demonstrated the allele-specific enhancer activity of this risk region using reporter assays. In UM, we identified the functional variant rs452384, of which the C risk allele is associated with higher gene expression, increased CLPTM1L expression in UM tumors, and a longer telomere length in peripheral blood mononuclear cells. Electrophoretic mobility shift assays and quantitative mass spectrometry identified NKX2.4 as an rs452384-T-specific binding protein, whereas GATA4 preferentially interacted with rs452384-C. Knockdown of NKX2.4 but not GATA4 resulted in increased TERT and CLPTM1L expression. In summary, the UM risk conferred by the 5p locus is at least partly due to rs452384, for which NKX2.4 presents strong differential binding activity and regulates CLPTM1L and TERT expression. Altogether, our work unraveled some of the complex regulatory mechanisms at the 5p15.33 susceptibility region in UM, and this might also shed light on shared mechanisms with other tumor types affected by this susceptibility region.
Keywords: TERT/CLPTM1L; cancer genetics; functional genomics; uveal melanoma.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures




References
-
- Jager M.J., Shields C.L., Cebulla C.M., Abdel-Rahman M.H., Grossniklaus H.E., Stern M.H., Carvajal R.D., Belfort R.N., Jia R., Shields J.A., Damato B.E. Uveal melanoma. Nat. Rev. Dis. Primers. 2020;6:24. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
