

Nascent peptide-induced translation discontinuation in eukaryotes impacts biased amino acid usage in proteomes
- PMID: 36460666
- PMCID: PMC9718836
- DOI: 10.1038/s41467-022-35156-x
Nascent peptide-induced translation discontinuation in eukaryotes impacts biased amino acid usage in proteomes
Abstract
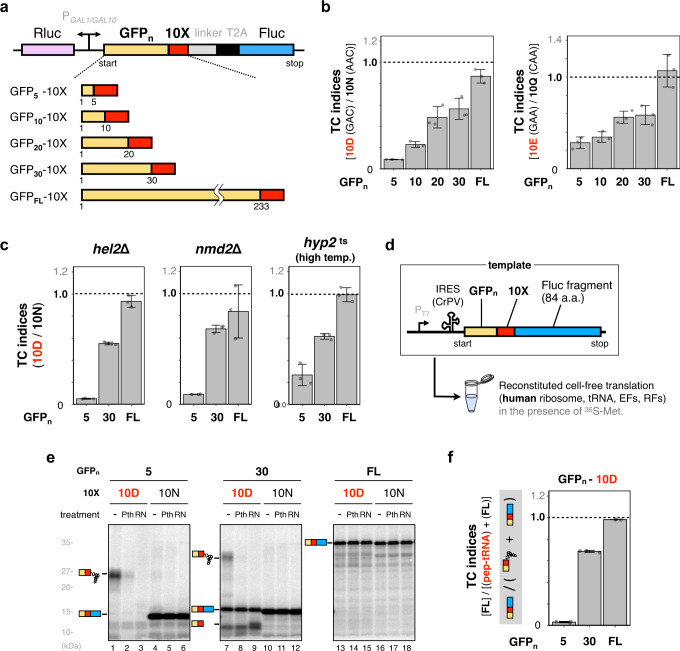
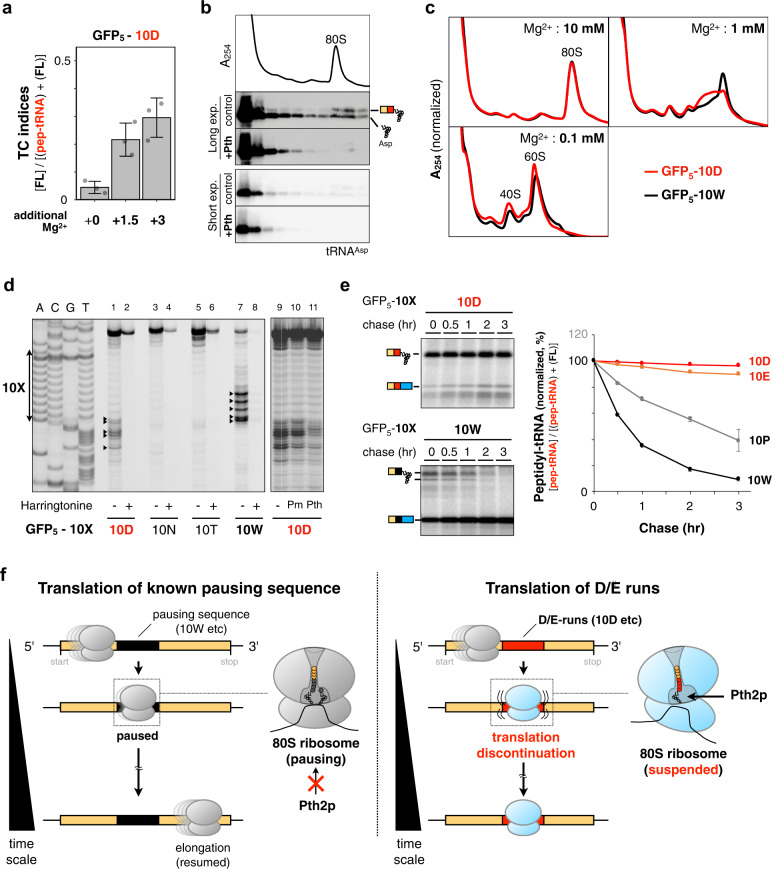
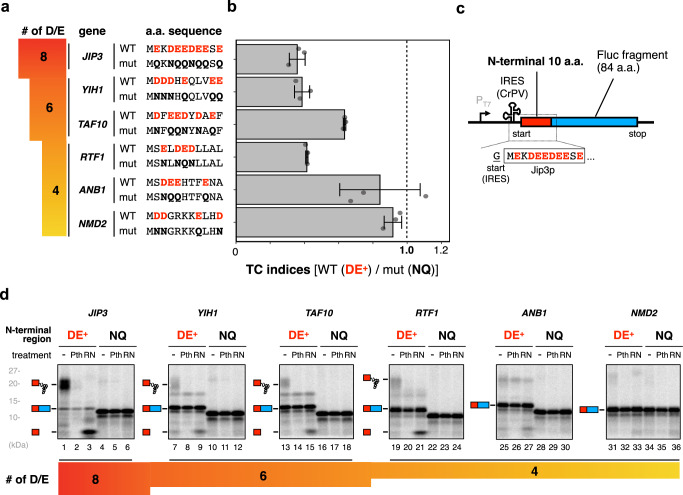
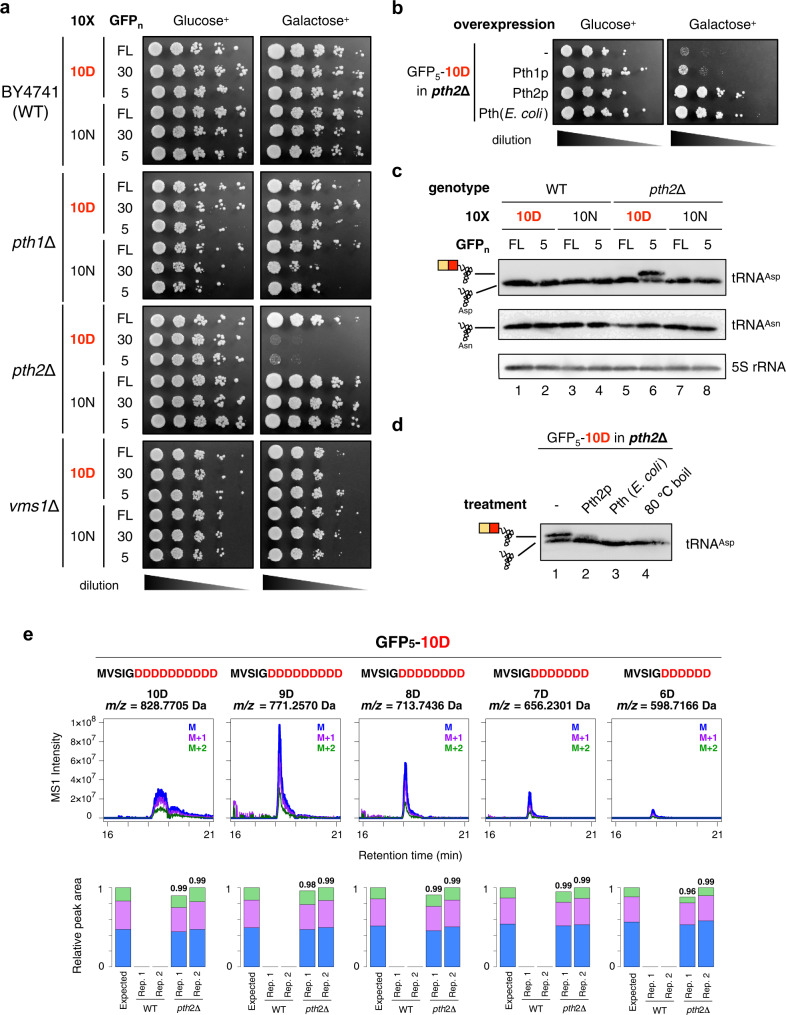
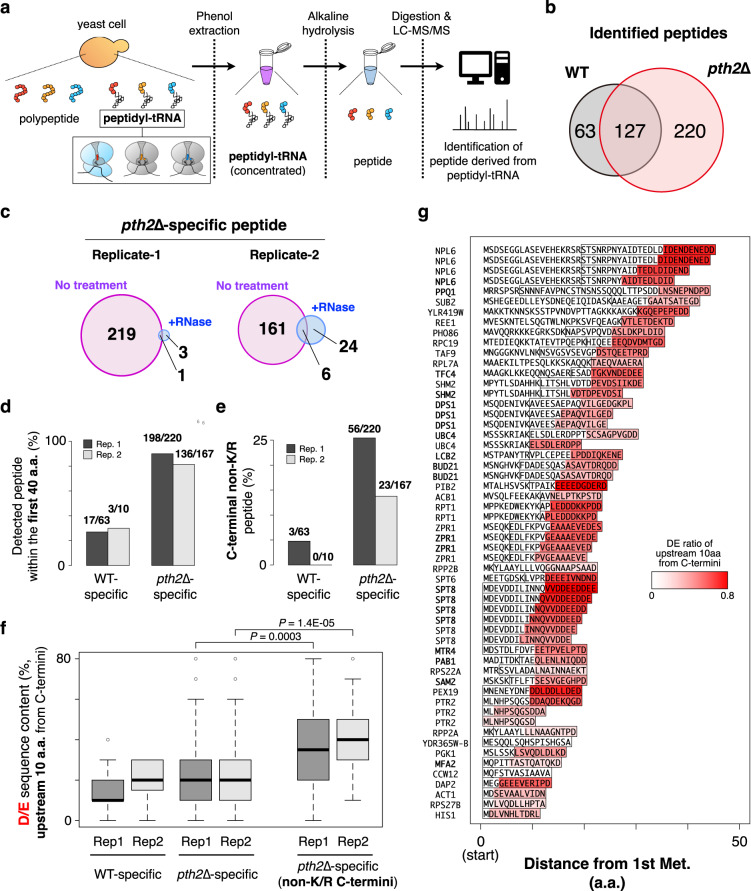
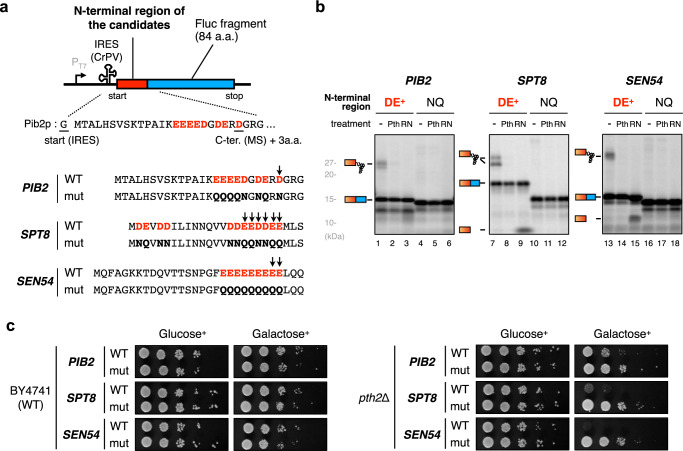
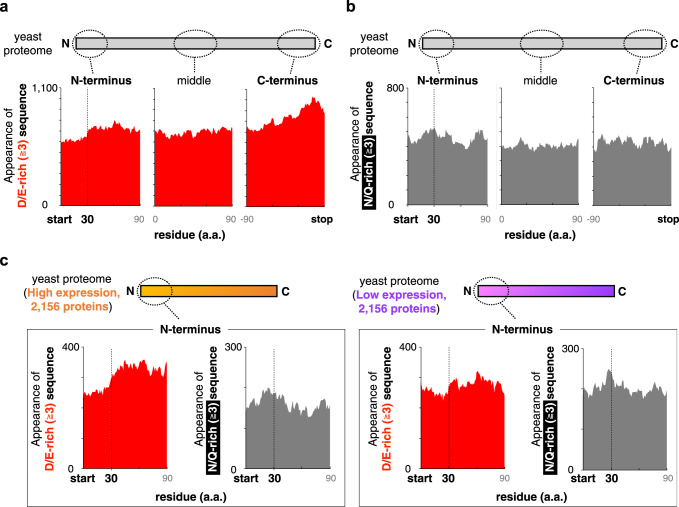
Robust translation elongation of any given amino acid sequence is required to shape proteomes. Nevertheless, nascent peptides occasionally destabilize ribosomes, since consecutive negatively charged residues in bacterial nascent chains can stochastically induce discontinuation of translation, in a phenomenon termed intrinsic ribosome destabilization (IRD). Here, using budding yeast and a human factor-based reconstituted translation system, we show that IRD also occurs in eukaryotic translation. Nascent chains enriched in aspartic acid (D) or glutamic acid (E) in their N-terminal regions alter canonical ribosome dynamics, stochastically aborting translation. Although eukaryotic ribosomes are more robust to ensure uninterrupted translation, we find many endogenous D/E-rich peptidyl-tRNAs in the N-terminal regions in cells lacking a peptidyl-tRNA hydrolase, indicating that the translation of the N-terminal D/E-rich sequences poses an inherent risk of failure. Indeed, a bioinformatics analysis reveals that the N-terminal regions of ORFs lack D/E enrichment, implying that the translation defect partly restricts the overall amino acid usage in proteomes.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Mechanistic dissection of premature translation termination induced by acidic residues-enriched nascent peptide.Cell Rep. 2023 Dec 26;42(12):113569. doi: 10.1016/j.celrep.2023.113569. Epub 2023 Dec 9. Cell Rep. 2023. PMID: 38071619
-
Intrinsic Ribosome Destabilization Underlies Translation and Provides an Organism with a Strategy of Environmental Sensing.Mol Cell. 2017 Nov 2;68(3):528-539.e5. doi: 10.1016/j.molcel.2017.10.020. Mol Cell. 2017. PMID: 29100053
-
Nascent polypeptide within the exit tunnel stabilizes the ribosome to counteract risky translation.EMBO J. 2021 Dec 1;40(23):e108299. doi: 10.15252/embj.2021108299. Epub 2021 Oct 20. EMBO J. 2021. PMID: 34672004 Free PMC article.
-
Ribosome regulation by the nascent peptide.Microbiol Rev. 1996 Jun;60(2):366-85. doi: 10.1128/mr.60.2.366-385.1996. Microbiol Rev. 1996. PMID: 8801438 Free PMC article. Review.
-
Nascent chain-mediated translation regulation in bacteria: translation arrest and intrinsic ribosome destabilization.J Biochem. 2023 Mar 31;173(4):227-236. doi: 10.1093/jb/mvad007. J Biochem. 2023. PMID: 36722132 Review.
Cited by
-
AntigenBoost: enhanced mRNA-based antigen expression through rational amino acid substitution.Brief Bioinform. 2024 Sep 23;25(6):bbae468. doi: 10.1093/bib/bbae468. Brief Bioinform. 2024. PMID: 39400114 Free PMC article.
-
The ABCF proteins in Escherichia coli individually cope with 'hard-to-translate' nascent peptide sequences.Nucleic Acids Res. 2024 Jun 10;52(10):5825-5840. doi: 10.1093/nar/gkae309. Nucleic Acids Res. 2024. PMID: 38661232 Free PMC article.
-
Interpreting ribosome dynamics during mRNA translation.J Biol Chem. 2025 Jul 10;301(8):110469. doi: 10.1016/j.jbc.2025.110469. Online ahead of print. J Biol Chem. 2025. PMID: 40651611 Free PMC article. Review.
-
Pyruvate Kinase M (PKM) binds ribosomes in a poly-ADP ribosylation dependent manner to induce translational stalling.Nucleic Acids Res. 2023 Jul 7;51(12):6461-6478. doi: 10.1093/nar/gkad440. Nucleic Acids Res. 2023. PMID: 37224531 Free PMC article.
-
Making Proteins with Electricity.Rev Physiol Biochem Pharmacol. 2025;187:195-237. doi: 10.1007/978-3-031-68827-0_13. Rev Physiol Biochem Pharmacol. 2025. PMID: 39838014 Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
