

Genetic epidemiology of inherited retinal diseases in a large patient cohort followed at a single center in Italy
- PMID: 36460718
- PMCID: PMC9718770
- DOI: 10.1038/s41598-022-24636-1
Genetic epidemiology of inherited retinal diseases in a large patient cohort followed at a single center in Italy
Abstract
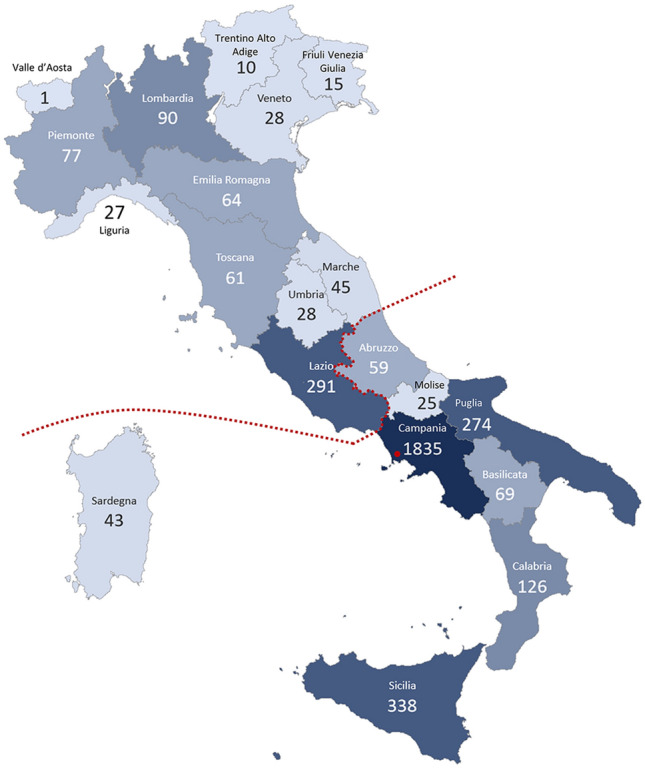
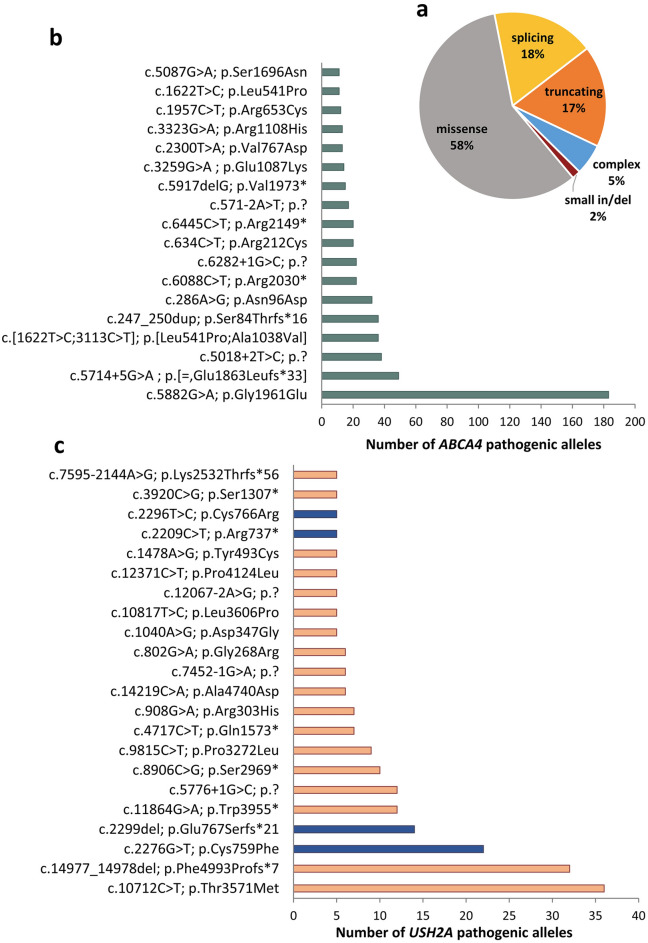
Inherited retinal diseases (IRDs) are the leading cause of vision loss in the working-age population. We performed a retrospective epidemiological study to determine the genetic basis of IRDs in a large Italian cohort (n = 2790) followed at a single referral center. We provided, mainly by next generation sequencing, potentially conclusive molecular diagnosis for 2036 patients (from 1683 unrelated families). We identified a total of 1319 causative sequence variations in 132 genes, including 353 novel variants, and 866 possibly actionable genotypes for therapeutic approaches. ABCA4 was the most frequently mutated gene (n = 535; 26.3% of solved cases), followed by USH2A (n = 228; 11.2%) and RPGR (n = 102; 5.01%). The other 129 genes had a lower contribution to IRD pathogenesis (e.g. CHM 3.5%, RHO 3.5%; MYO7A 3.4%; CRB1 2.7%; RPE65 2%, RP1 1.8%; GUCY2D 1.7%). Seventy-eight genes were mutated in five patients or less. Mitochondrial DNA variants were responsible for 2.1% of cases. Our analysis confirms the complex genetic etiology of IRDs and reveals the high prevalence of ABCA4 and USH2A mutations. This study also uncovers genetic associations with a spectrum of clinical subgroups and highlights a valuable number of cases potentially eligible for clinical trials and, ultimately, for molecular therapies.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
