

CAULIFINDER: a pipeline for the automated detection and annotation of caulimovirid endogenous viral elements in plant genomes
- PMID: 36463202
- PMCID: PMC9719215
- DOI: 10.1186/s13100-022-00288-w
CAULIFINDER: a pipeline for the automated detection and annotation of caulimovirid endogenous viral elements in plant genomes
Abstract
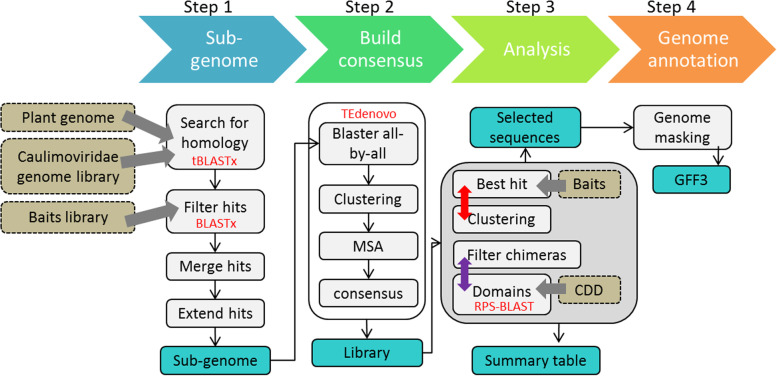
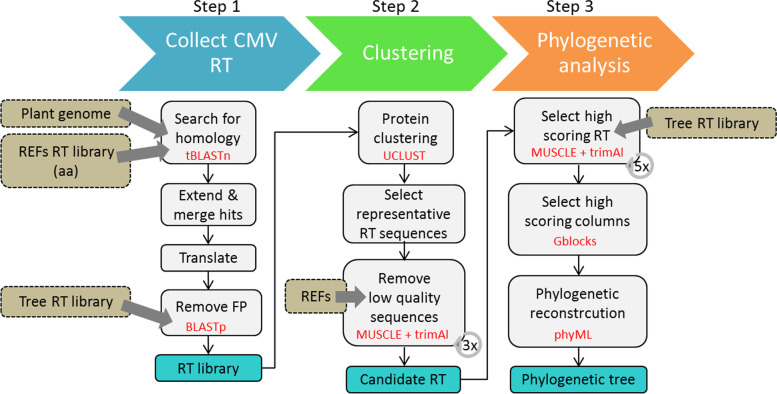
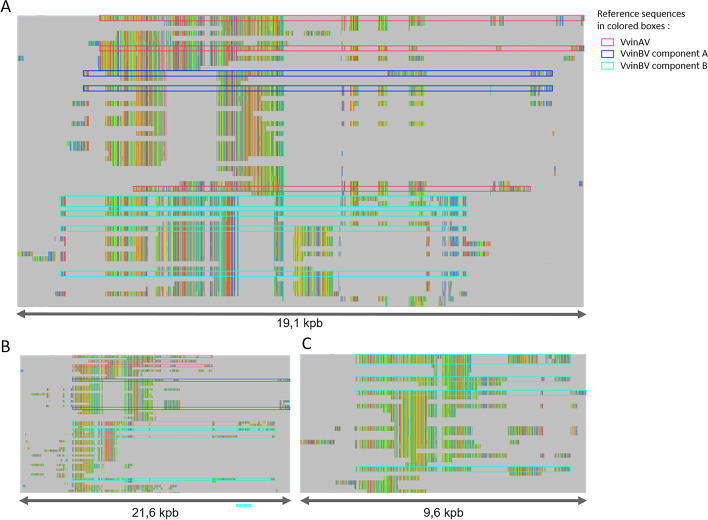
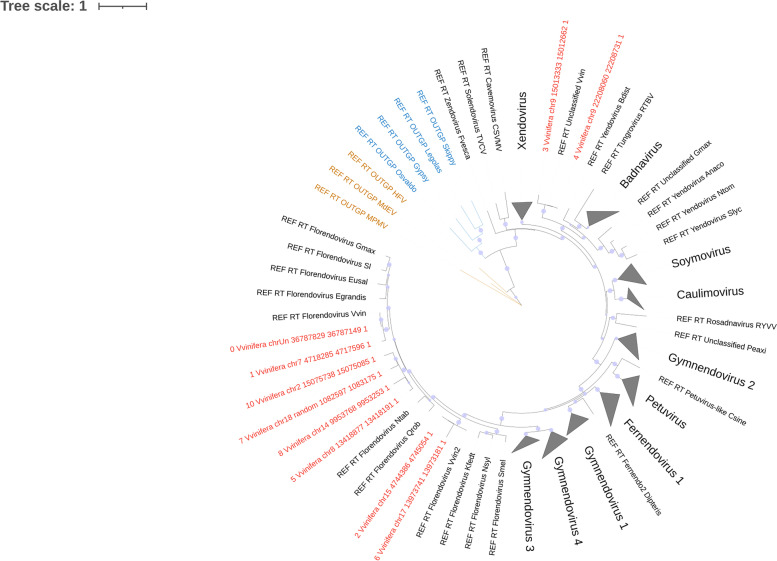
Plant, animal and protist genomes often contain endogenous viral elements (EVEs), which correspond to partial and sometimes entire viral genomes that have been captured in the genome of their host organism through a variety of integration mechanisms. While the number of sequenced eukaryotic genomes is rapidly increasing, the annotation and characterization of EVEs remains largely overlooked. EVEs that derive from members of the family Caulimoviridae are widespread across tracheophyte plants, and sometimes they occur in very high copy numbers. However, existing programs for annotating repetitive DNA elements in plant genomes are poor at identifying and then classifying these EVEs. Other than accurately annotating plant genomes, there is intrinsic value in a tool that could identify caulimovirid EVEs as they testify to recent or ancient host-virus interactions and provide valuable insights into virus evolution. In response to this research need, we have developed CAULIFINDER, an automated and sensitive annotation software package. CAULIFINDER consists of two complementary workflows, one to reconstruct, annotate and group caulimovirid EVEs in a given plant genome and the second to classify these genetic elements into officially recognized or tentative genera in the Caulimoviridae. We have benchmarked the CAULIFINDER package using the Vitis vinifera reference genome, which contains a rich assortment of caulimovirid EVEs that have previously been characterized using manual methods. The CAULIFINDER package is distributed in the form of a Docker image.
Keywords: Bioinformatics; Caulimoviridae; Endogenous viral elements; Genome annotation; Paleovirology; Plant genomes; Repetitive elements.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
