

NS1619 Alleviate Brain-Derived Extracellular Vesicle-Induced Brain Injury by Regulating BKca Channel and Nrf2/HO-1/NF-ĸB Pathway
- PMID: 36466093
- PMCID: PMC9711983
- DOI: 10.1155/2022/2257427
NS1619 Alleviate Brain-Derived Extracellular Vesicle-Induced Brain Injury by Regulating BKca Channel and Nrf2/HO-1/NF-ĸB Pathway
Abstract
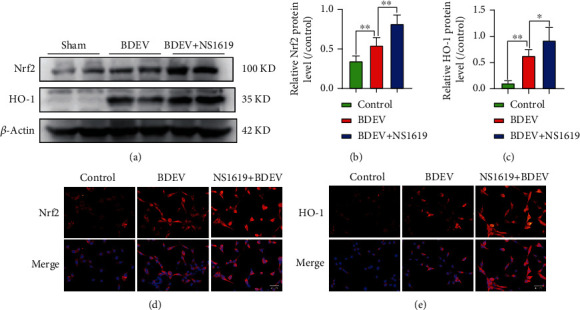
Brain induced extracellular vesicle (BDEV) elevates after traumatic brain injury (TBI) and contributes to secondary brain injury. However, the role of BDEV in TBI remains unclear. In this study, we determined the mechanisms of BDEV in brain injury and explored whether neuroprotective drug BKca channel opener NS1619 may attenuate BDEV-induced brain injury. We injected BDEV and lactadherin, respectively, to mimic the up and downregulation of BDEV after TBI and illustrated the role of BDEV in vivo. In vitro, the membrane potential and calcium concentration of HT-22, bEnd3, and BV-2 were measured by fluorescent staining. The effects of BDEV and NS1619 on HT-22 were evaluated by CCK-8, LDH release assay, Na+/k+-ATPase activity, JC-1 staining, DHE staining, and 4-HNE staining, respectively. The role of BDEV and NS1619 on the Nrf2/HO-1/p65 pathway was also evaluated in HT-22. Finally, we administrated TBI mice with NS1619 to clarify the role of NS1619 against BDEV in vivo. Our results suggested that BDEV aggravated and lactadherin mitigated TBI-induced EB leakage, brain edema, neuronal degeneration, apoptosis, ROS level, microgliosis, MMP-9 activity, and NF-κB activation. In vitro, BDEV-caused depolarized membrane potential and calcium overload were significantly attenuated by NS1619 in HT-22, bEnd3, and BV-2. BDEV markedly decreased cell viability, Na+/k+-ATPase activity, and caused mitochondrial dysregulation, oxidative stress, and NF-ĸB activation. NS1619 pretreatment alleviated above process and enhanced antioxidant system Nrf2/HO-1 in HT-22. Finally, NS1619 administration significantly inhibited neuroinflammation response and improved TBI outcome after TBI. NS1619 treatment also reduced 4-HNE content and NF-ĸB activation and enhanced Nrf2/HO-1 pathway. Our data showed that BDEV aggravated brain injury by perturbing cell membrane potential, calcium homeostasis, oxidative stress, and neuroinflammation. The BKca channel opener NS1619 attenuated BDEV-induced pathological process in vitro and in vivo by modulating the BKca channel and Nrf2/HO-1/NF-ĸB pathway.
Copyright © 2022 Yalong Gao et al.
Conflict of interest statement
These authors declare that they have no competing interests.
Figures










Similar articles
-
Traumatic brain injury-induced downregulation of Nrf2 activates inflammatory response and apoptotic cell death.J Mol Med (Berl). 2019 Dec;97(12):1627-1641. doi: 10.1007/s00109-019-01851-4. Epub 2019 Nov 22. J Mol Med (Berl). 2019. PMID: 31758217
-
Annexin A5 ameliorates traumatic brain injury-induced neuroinflammation and neuronal ferroptosis by modulating the NF-ĸB/HMGB1 and Nrf2/HO-1 pathways.Int Immunopharmacol. 2023 Jan;114:109619. doi: 10.1016/j.intimp.2022.109619. Epub 2022 Dec 21. Int Immunopharmacol. 2023. PMID: 36700781
-
N-acetylcysteine amide provides neuroprotection via Nrf2-ARE pathway in a mouse model of traumatic brain injury.Drug Des Devel Ther. 2018 Dec 4;12:4117-4127. doi: 10.2147/DDDT.S179227. eCollection 2018. Drug Des Devel Ther. 2018. PMID: 30584276 Free PMC article.
-
Targeting Nrf2-Mediated Oxidative Stress Response in Traumatic Brain Injury: Therapeutic Perspectives of Phytochemicals.Oxid Med Cell Longev. 2022 Apr 4;2022:1015791. doi: 10.1155/2022/1015791. eCollection 2022. Oxid Med Cell Longev. 2022. PMID: 35419162 Free PMC article. Review.
-
Targeting the NF-E2-Related Factor 2 Pathway: a Novel Strategy for Traumatic Brain Injury.Mol Neurobiol. 2018 Feb;55(2):1773-1785. doi: 10.1007/s12035-017-0456-z. Epub 2017 Feb 21. Mol Neurobiol. 2018. PMID: 28224478 Review.
Cited by
-
Neuroprotection in an Experimental Model of Multiple Sclerosis via Opening of Big Conductance, Calcium-Activated Potassium Channels.Pharmaceuticals (Basel). 2023 Jul 7;16(7):972. doi: 10.3390/ph16070972. Pharmaceuticals (Basel). 2023. PMID: 37513884 Free PMC article.
-
Oxidative State in Cutaneous Melanoma Progression: A Question of Balance.Antioxidants (Basel). 2024 Aug 30;13(9):1058. doi: 10.3390/antiox13091058. Antioxidants (Basel). 2024. PMID: 39334716 Free PMC article. Review.
-
3,3'-Diindolylmethane improves pathology and neurological outcome following traumatic brain injury.Neurotherapeutics. 2025 Mar;22(2):e00531. doi: 10.1016/j.neurot.2025.e00531. Epub 2025 Feb 4. Neurotherapeutics. 2025. PMID: 39909809 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
