

Good practices and recommendations for using and benchmarking computational metabolomics metabolite annotation tools
- PMID: 36469190
- PMCID: PMC9722809
- DOI: 10.1007/s11306-022-01963-y
Good practices and recommendations for using and benchmarking computational metabolomics metabolite annotation tools
Abstract
Background: Untargeted metabolomics approaches based on mass spectrometry obtain comprehensive profiles of complex biological samples. However, on average only 10% of the molecules can be annotated. This low annotation rate hampers biochemical interpretation and effective comparison of metabolomics studies. Furthermore, de novo structural characterization of mass spectral data remains a complicated and time-intensive process. Recently, the field of computational metabolomics has gained traction and novel methods have started to enable large-scale and reliable metabolite annotation. Molecular networking and machine learning-based in-silico annotation tools have been shown to greatly assist metabolite characterization in diverse fields such as clinical metabolomics and natural product discovery.
Aim of review: We highlight recent advances in computational metabolite annotation workflows with a special focus on their evaluation and comparison with other tools. Whilst the progress is substantial and promising, we also argue that inconsistencies in benchmarking different tools hamper users from selecting the most appropriate and promising method for their research. We summarize benchmarking strategies of the different tools and outline several recommendations for benchmarking and comparing novel tools.
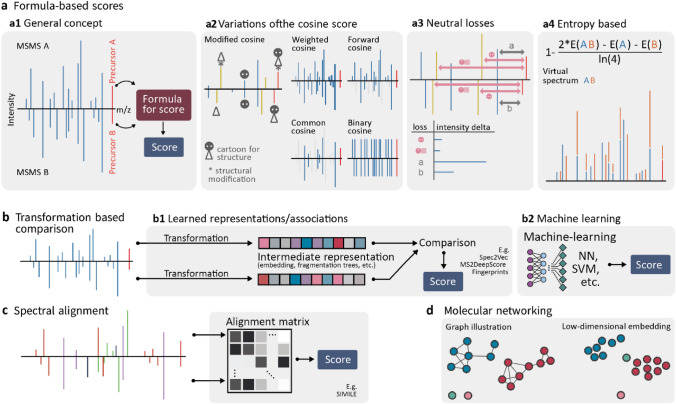
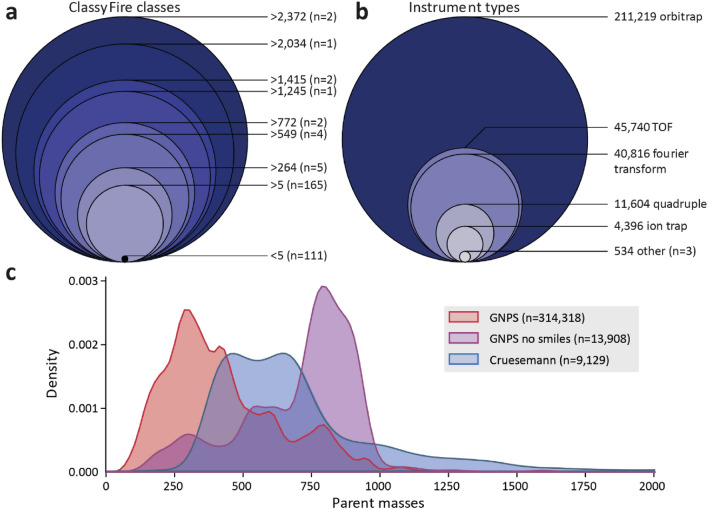
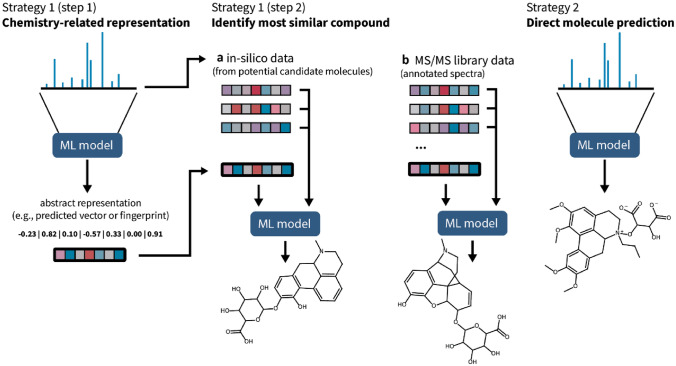
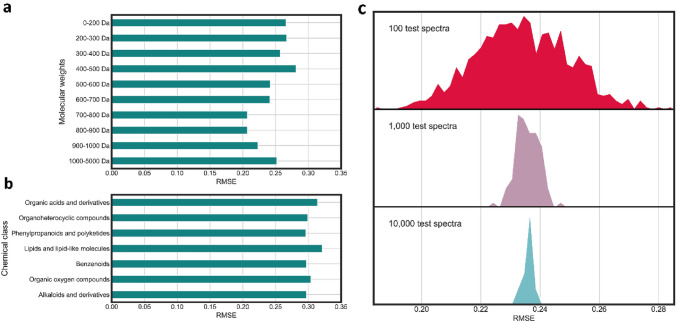
Key scientific concepts of review: This review focuses on recent advances in mass spectral library-based and machine learning-supported metabolite annotation workflows. We discuss large-scale library matching and analogue search, the current bloom of mass spectral similarity scores, and how molecular networking has changed the field. In addition, the potentials and challenges of machine learning-supported metabolite annotation workflows are highlighted. Overall, recent developments in computational metabolomics have started to fundamentally change metabolomics workflows, and we expect that as a community we will be able to overcome current method performance ambiguities and annotation bottlenecks.
Keywords: Benchmarking; Machine learning; Mass fragmentation spectra; Mass spectrometry; Metabolite annotation and identification; Untargeted metabolomics.
© 2022. The Author(s).
Conflict of interest statement
Justin J.J. van der Hooft is a member of the Scientific Advisory Board of NAICONS Srl, Milano, Italy. All other authors declare no conflict of interests.
Figures
References
-
- Alseekh S, Aharoni A, Brotman Y, Contrepois K, D'Auria J, Ewald J, Fraser PD, Giavalisco P, Hall RD, Heinemann M, Link H, Luo J, Neumann S, Nielsen J, Perez de Souza L, Saito K, Sauer U, Schroeder FC, Schuster S, et al. Mass spectrometry-based metabolomics: a guide for annotation, quantification and best reporting practices. Natural Methods. 2021;18:747–756. doi: 10.1038/s41592-021-01197-1. - DOI - PMC - PubMed
-
- Aron AT, Gentry EC, McPhail KL, Nothias L-F, Nothias-Esposito M, Bouslimani A, Petras D, Gauglitz JM, Sikora N, Vargas F, van der Hooft JJJ, Ernst M, Kang KB, Aceves CM, Caraballo-Rodríguez AM, Koester I, Weldon KC, Bertrand S, Roullier C, et al. Reproducible molecular networking of untargeted mass spectrometry data using GNPS. Nature Protocols. 2020;15:1954–1991. doi: 10.1038/s41596-020-0317-5. - DOI - PubMed
-
- Bach, E., Schymanski, E. L., & Rousu, J. (2022) Joint structural annotation of small molecules using liquid chromatography retention order and tandem mass spectrometry data. bioRxiv.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials