

JCAD promotes arterial thrombosis through PI3K/Akt modulation: a translational study
- PMID: 36469488
- PMCID: PMC10200023
- DOI: 10.1093/eurheartj/ehac641
JCAD promotes arterial thrombosis through PI3K/Akt modulation: a translational study
Erratum in
-
Corrigendum to: JCAD promotes arterial thrombosis through PI3K/Akt modulation: a translational study.Eur Heart J. 2023 May 21;44(20):1843. doi: 10.1093/eurheartj/ehad174. Eur Heart J. 2023. PMID: 37171028 Free PMC article. No abstract available.
Abstract
Aims: Variants of the junctional cadherin 5 associated (JCAD) locus associate with acute coronary syndromes. JCAD promotes experimental atherosclerosis through the large tumor suppressor kinase 2 (LATS2)/Hippo pathway. This study investigates the role of JCAD in arterial thrombosis.
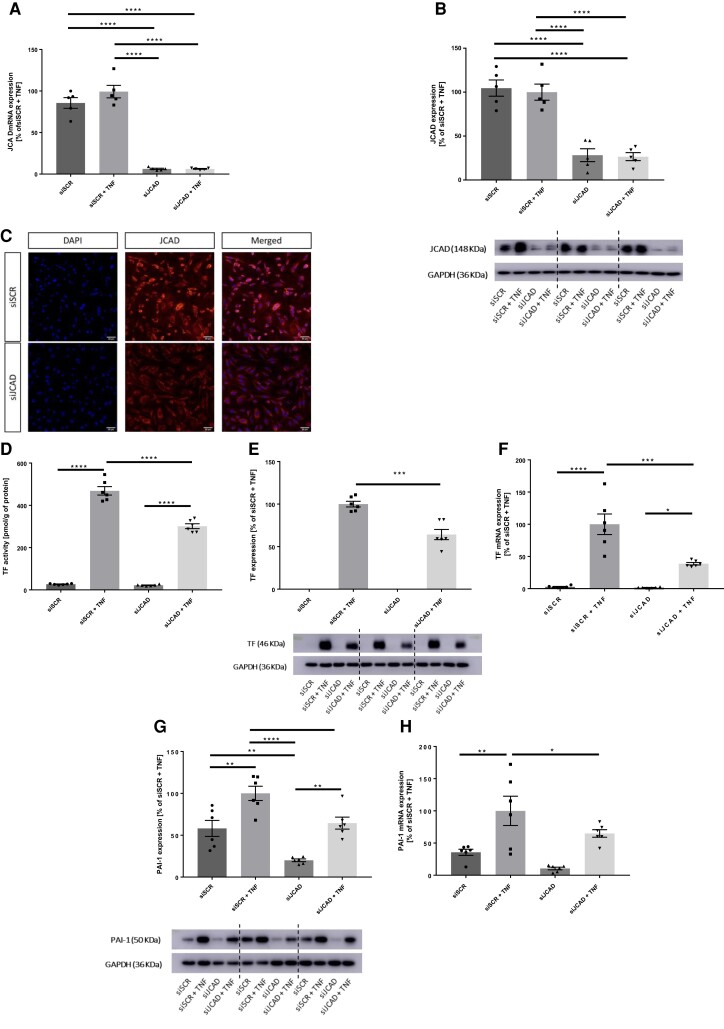
Methods and results: JCAD knockout (Jcad-/-) mice underwent photochemically induced endothelial injury to trigger arterial thrombosis. Primary human aortic endothelial cells (HAECs) treated with JCAD small interfering RNA (siJCAD), LATS2 small interfering RNA (siLATS2) or control siRNA (siSCR) were employed for in vitro assays. Plasma JCAD was measured in patients with chronic coronary syndrome or ST-elevation myocardial infarction (STEMI). Jcad-/- mice displayed reduced thrombogenicity as reflected by delayed time to carotid occlusion. Mechanisms include reduced activation of the coagulation cascade [reduced tissue factor (TF) expression and activity] and increased fibrinolysis [higher thrombus embolization episodes and D-dimer levels, reduced vascular plasminogen activator inhibitor (PAI)-1 expression]. In vitro, JCAD silencing inhibited TF and PAI-1 expression in HAECs. JCAD-silenced HAECs (siJCAD) displayed increased levels of LATS2 kinase. Yet, double JCAD and LATS2 silencing did not restore the control phenotype. si-JCAD HAECs showed increased levels of phosphoinositide 3-kinases (PI3K)/ proteinkinase B (Akt) activation, known to downregulate procoagulant expression. The PI3K/Akt pathway inhibitor-wortmannin-prevented the effect of JCAD silencing on TF and PAI-1, indicating a causative role. Also, co-immunoprecipitation unveiled a direct interaction between JCAD and Akt. Confirming in vitro findings, PI3K/Akt and P-yes-associated protein levels were higher in Jcad-/- animals. Lastly, as compared with chronic coronary syndrome, STEMI patients showed higher plasma JCAD, which notably correlated positively with both TF and PAI-1 levels.
Conclusions: JCAD promotes arterial thrombosis by modulating coagulation and fibrinolysis. Herein, reported translational data suggest JCAD as a potential therapeutic target for atherothrombosis.
Keywords: Arterial thrombosis; Cardiovascular disease; JCAD; KIAA1462; PAI-1; Tissue factor.
© The Author(s) 2022. Published by Oxford University Press on behalf of the European Society of Cardiology. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Conflict of interest statement
Conflict of interest: L.L. and G.G.C. are coinventors on the International Patent WO/2020/226993 filed in April 2020. The patent relates to the use of antibodies which specifically bind IL-1α to reduce various sequelae of ischemia-reperfusion injury to the central nervous system. G.G.C. is a consultant to Sovida solutions limited. T.F.L. has no conflicts related to this manuscript. Outside the work he received unrestricted research and education grants from Abbott, Amgen, Boehringer Ingelheim, Daichi-Sankyo, Novartis, Roche Diagnostics, Sanofi, Servier and Vifor. L.L. reports speaker fees outside of this work from Daichi-Sankyo. G.L. reports Consulting fees and Payment or honoraria for lectures: Novo Nordisk, Novartis, Sanofi, AstraZeneca, Boehringer, Bayer. F.C. reports speaker fees from Amgen, Astra Zeneca, Servier, BMS, other from GlyCardial Diagnostics, outside the submitted work. The other authors report no conflict of interest.
Figures








Comment in
-
JCAD: a new GWAS target to reduce residual cardiovascular risk?Eur Heart J. 2023 May 21;44(20):1834-1836. doi: 10.1093/eurheartj/ehac708. Eur Heart J. 2023. PMID: 36514298 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
