

Multiclonal human origin and global expansion of an endemic bacterial pathogen of livestock
- PMID: 36469788
- PMCID: PMC9897428
- DOI: 10.1073/pnas.2211217119
Multiclonal human origin and global expansion of an endemic bacterial pathogen of livestock
Abstract
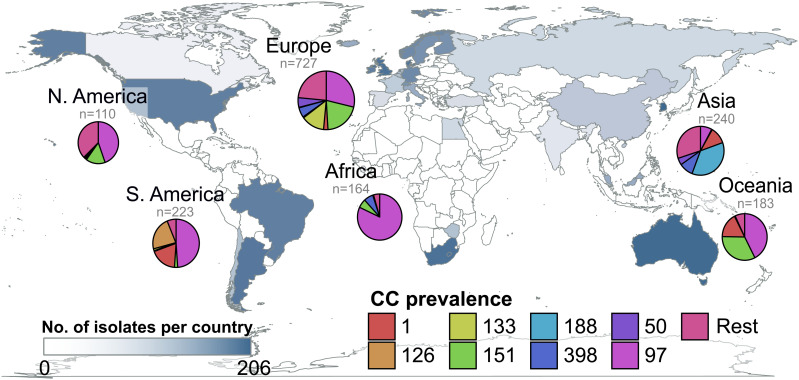
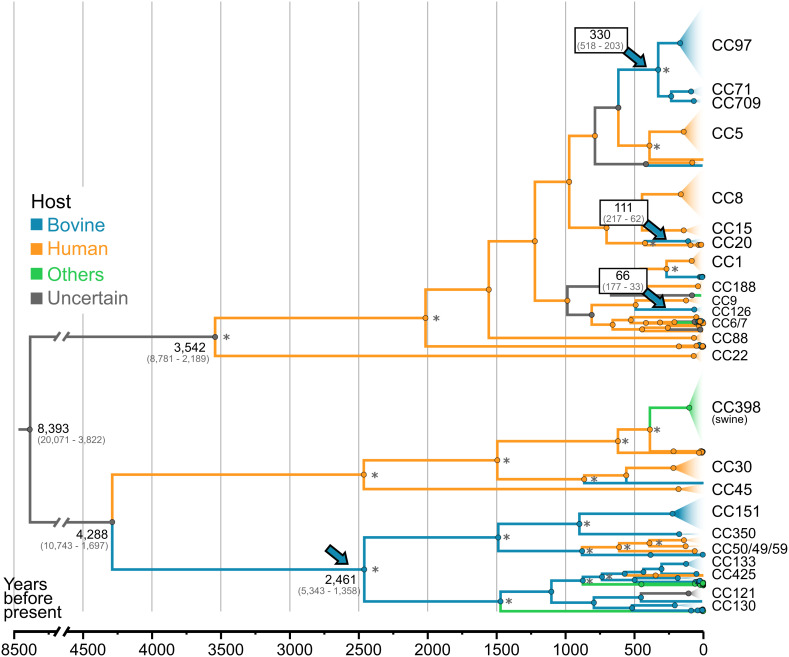
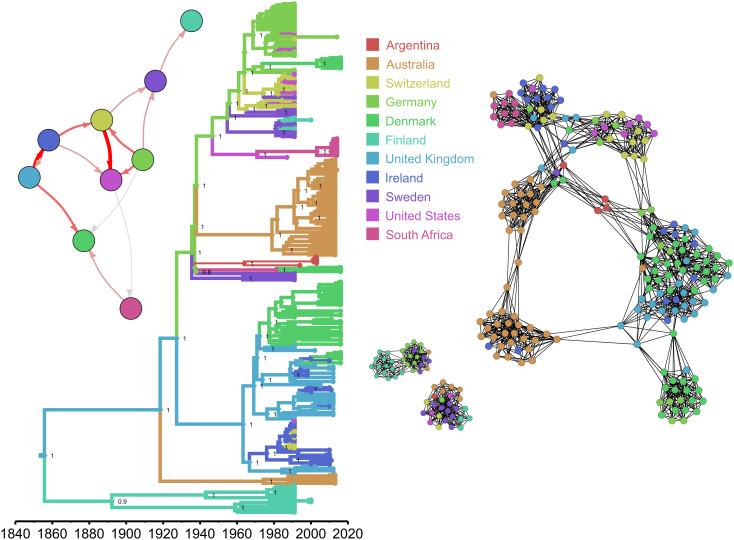
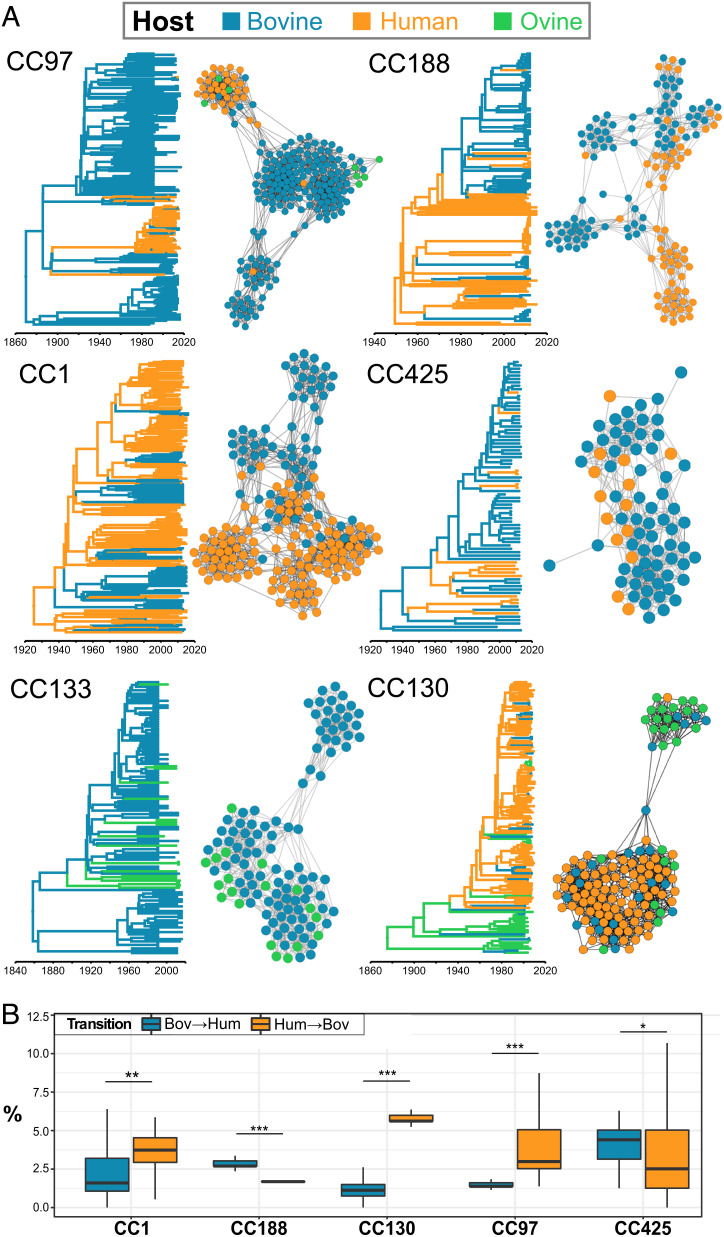
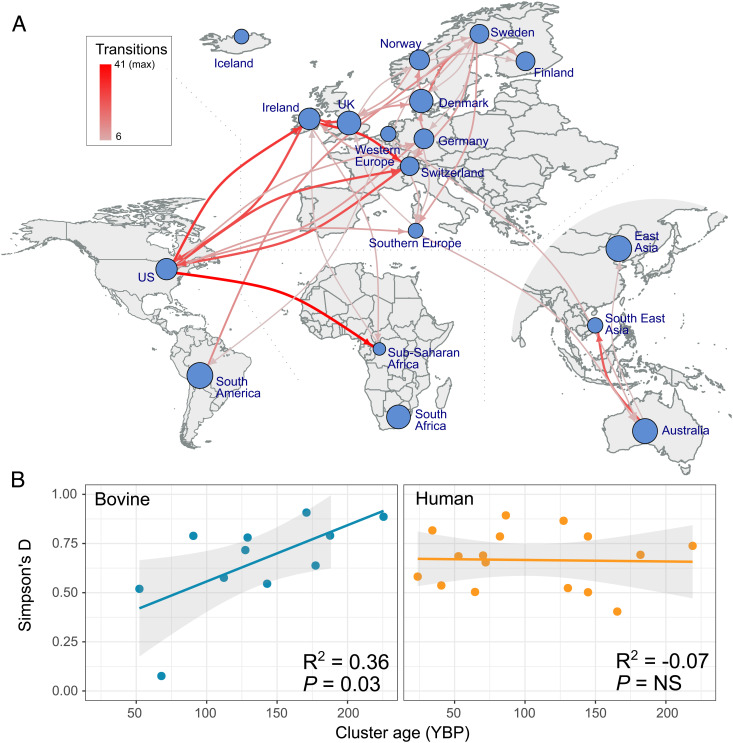
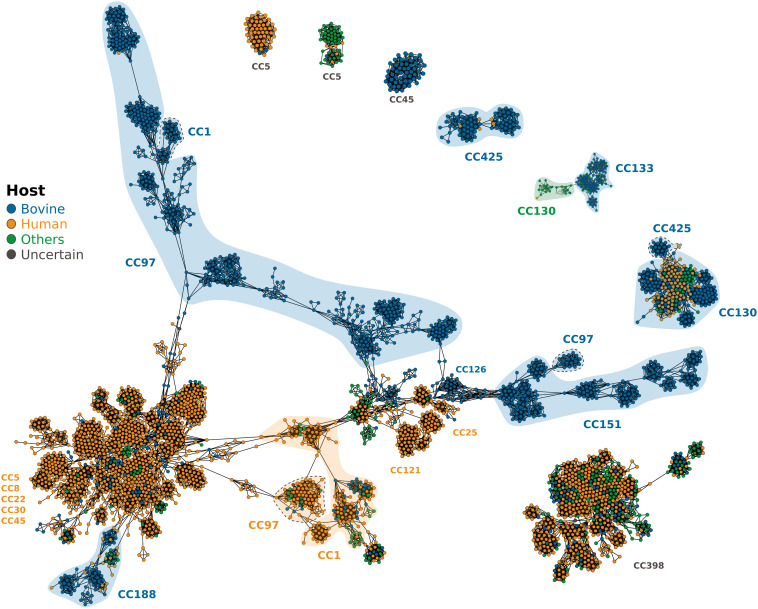
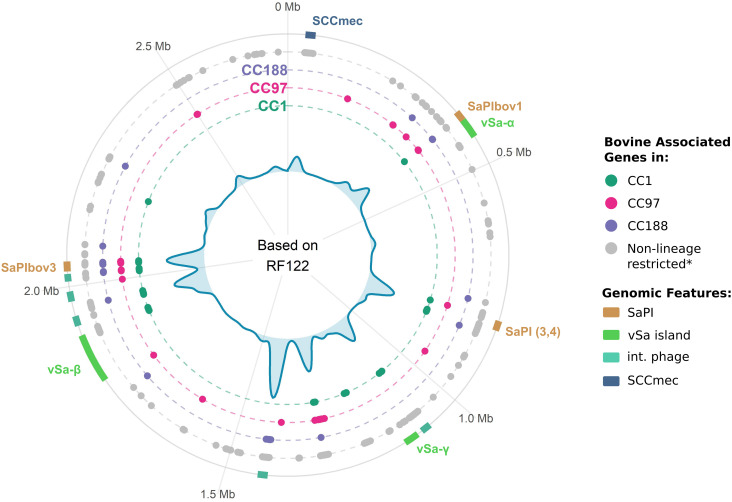
Most new pathogens of humans and animals arise via switching events from distinct host species. However, our understanding of the evolutionary and ecological drivers of successful host adaptation, expansion, and dissemination are limited. Staphylococcus aureus is a major bacterial pathogen of humans and a leading cause of mastitis in dairy cows worldwide. Here we trace the evolutionary history of bovine S. aureus using a global dataset of 10,254 S. aureus genomes including 1,896 bovine isolates from 32 countries in 6 continents. We identified 7 major contemporary endemic clones of S. aureus causing bovine mastitis around the world and traced them back to 4 independent host-jump events from humans that occurred up to 2,500 y ago. Individual clones emerged and underwent clonal expansion from the mid-19th to late 20th century coinciding with the commercialization and industrialization of dairy farming, and older lineages have become globally distributed via established cattle trade links. Importantly, we identified lineage-dependent differences in the frequency of host transmission events between humans and cows in both directions revealing high risk clones threatening veterinary and human health. Finally, pangenome network analysis revealed that some bovine S. aureus lineages contained distinct sets of bovine-associated genes, consistent with multiple trajectories to host adaptation via gene acquisition. Taken together, we have dissected the evolutionary history of a major endemic pathogen of livestock providing a comprehensive temporal, geographic, and gene-level perspective of its remarkable success.
Keywords: Staphylococcus aureus; agriculture; host adaptation; phylodynamics; population genomics.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Morand S., McIntyre K. M., Baylis M., Domesticated animals and human infectious diseases of zoonotic origins: Domestication time matters. Infect. Genet. Evol. 24, 76–81 (2014). - PubMed
-
- Peton V., Le Loir Y., Staphylococcus aureus in veterinary medicine. Infect. Genet. Evol. 21, 602–615 (2014). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
