

The landscape of tumor cell states and spatial organization in H3-K27M mutant diffuse midline glioma across age and location
- PMID: 36471067
- PMCID: PMC9729116
- DOI: 10.1038/s41588-022-01236-3
The landscape of tumor cell states and spatial organization in H3-K27M mutant diffuse midline glioma across age and location
Abstract
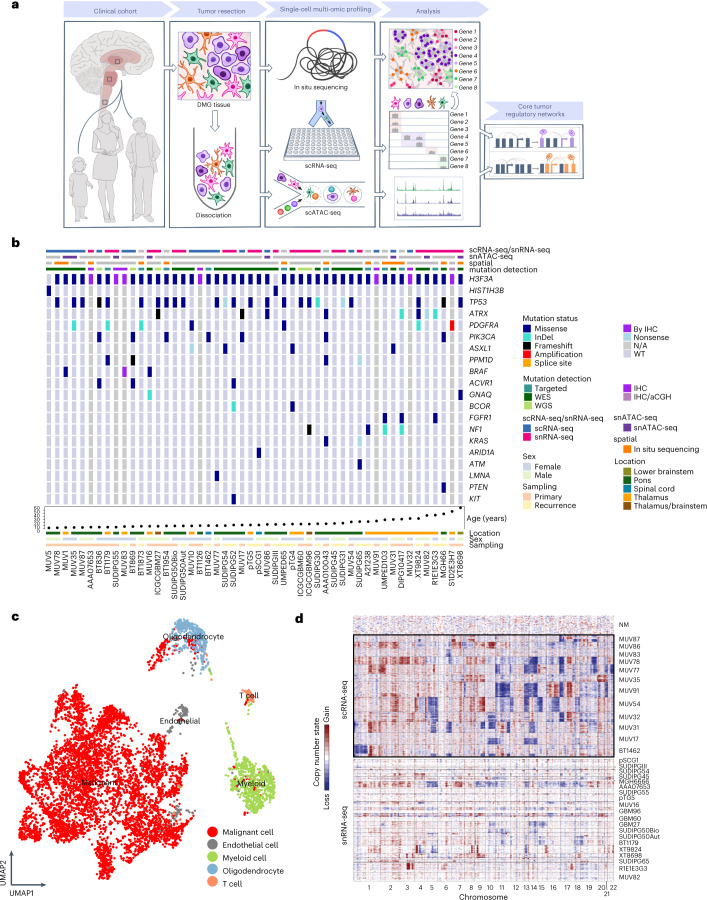
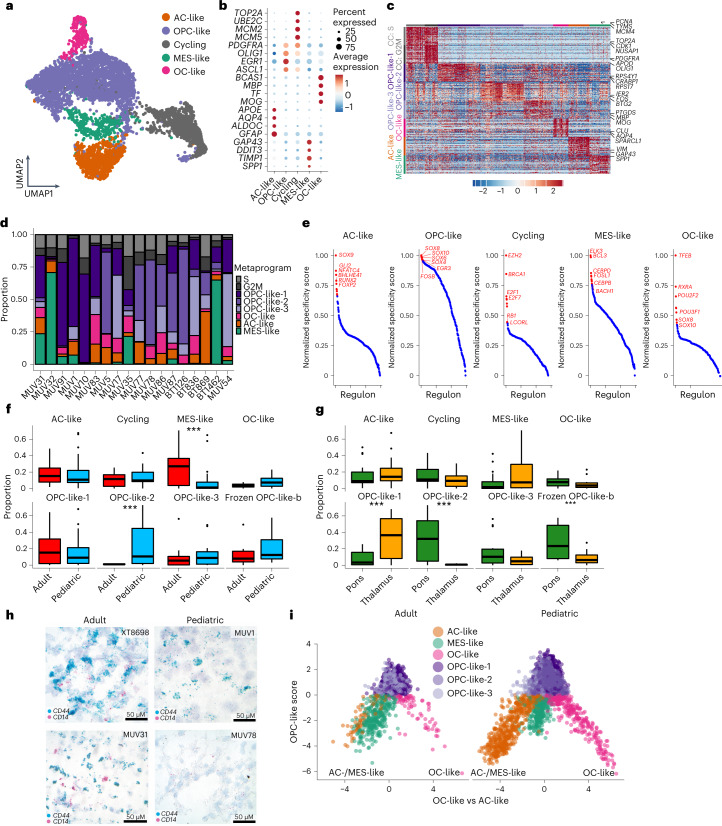
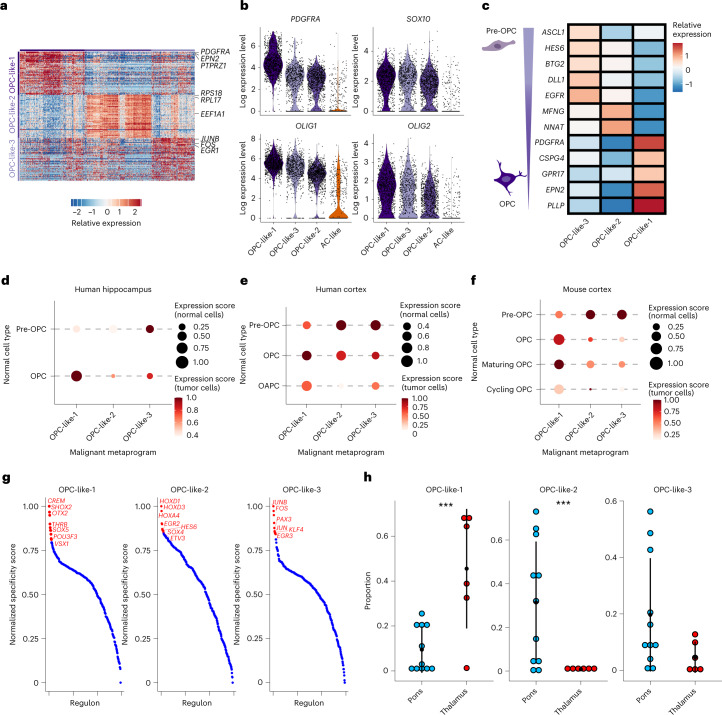
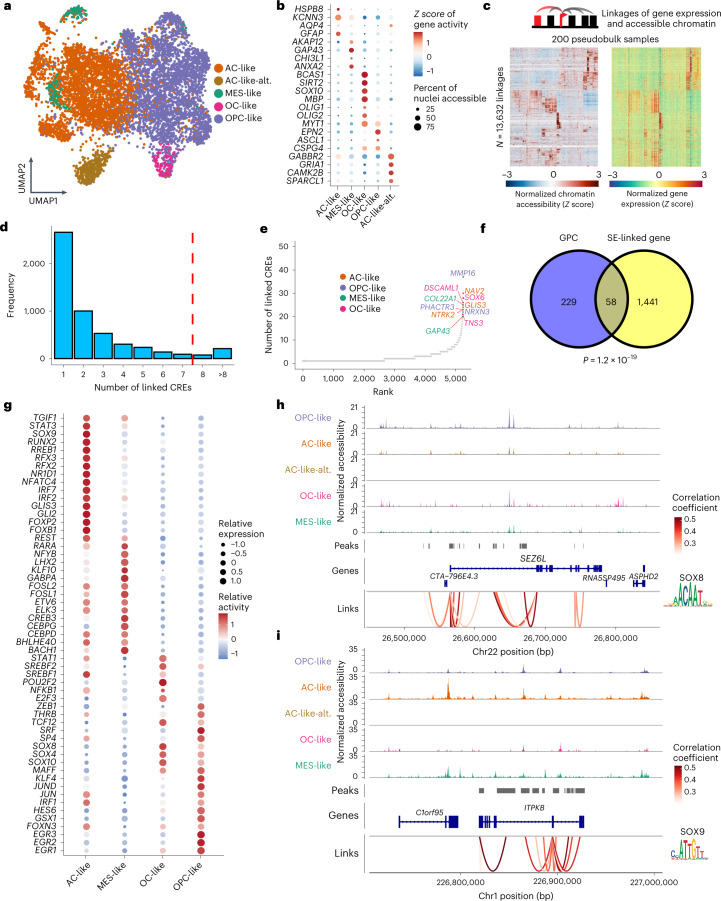
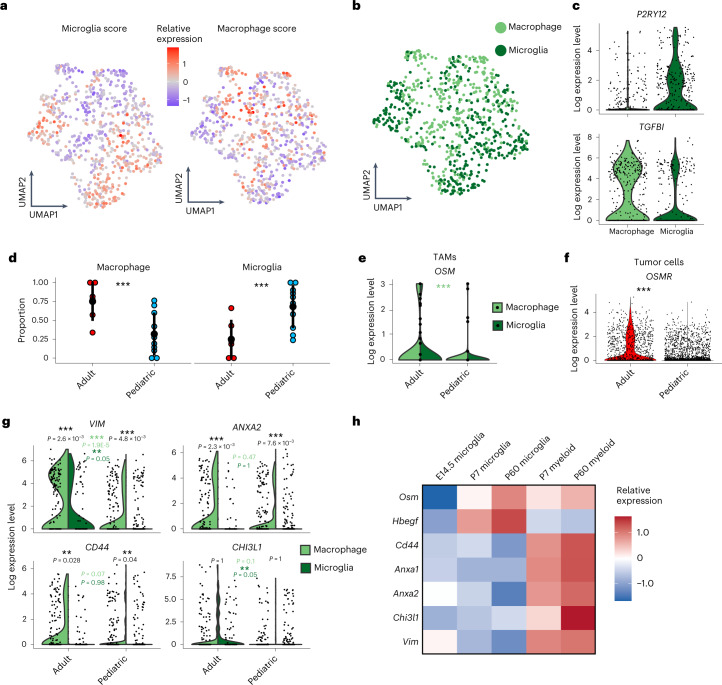
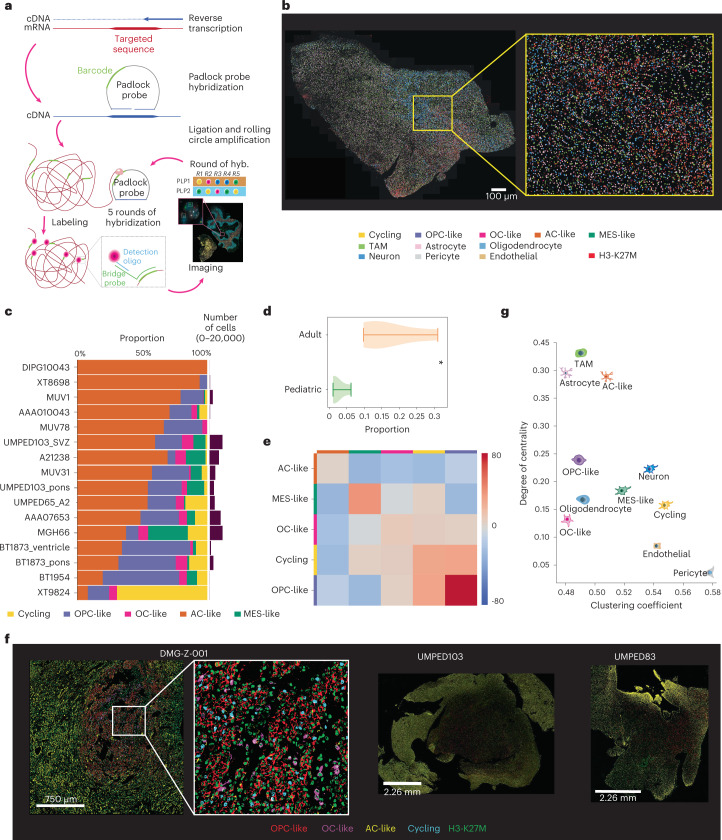
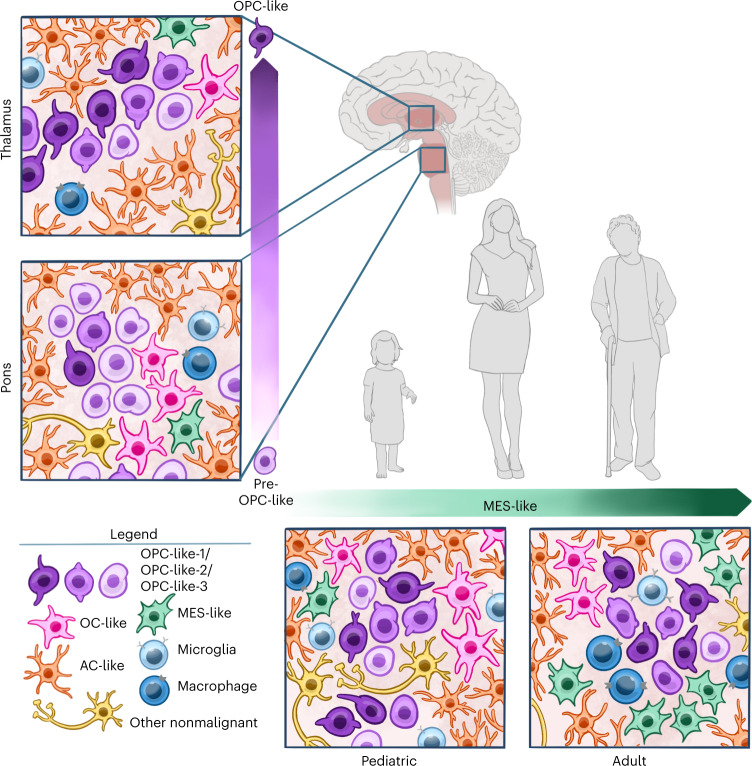
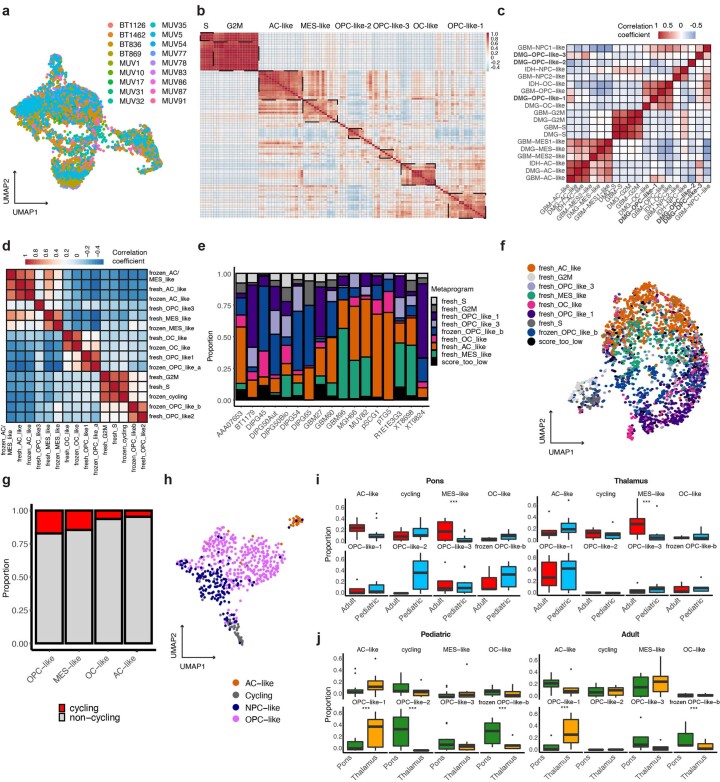
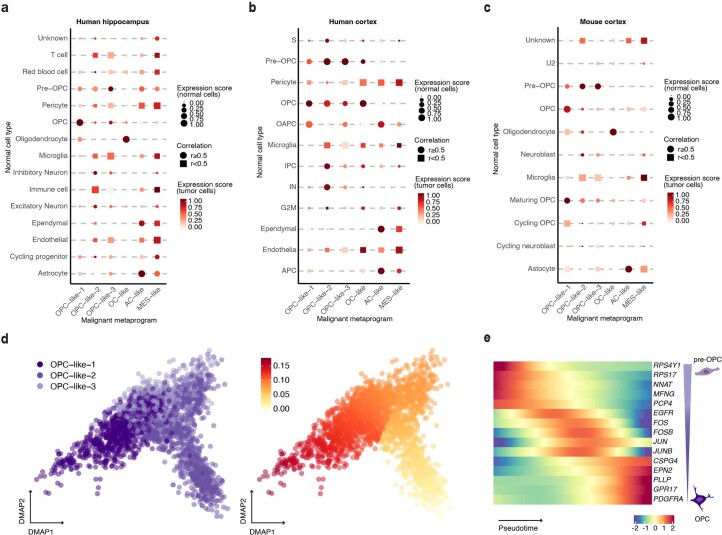
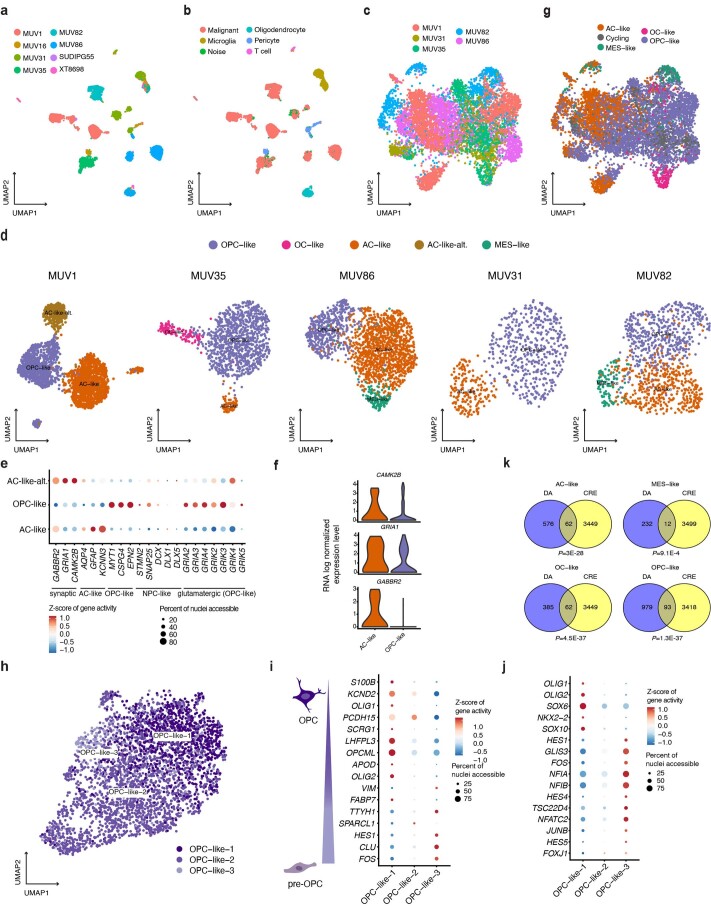
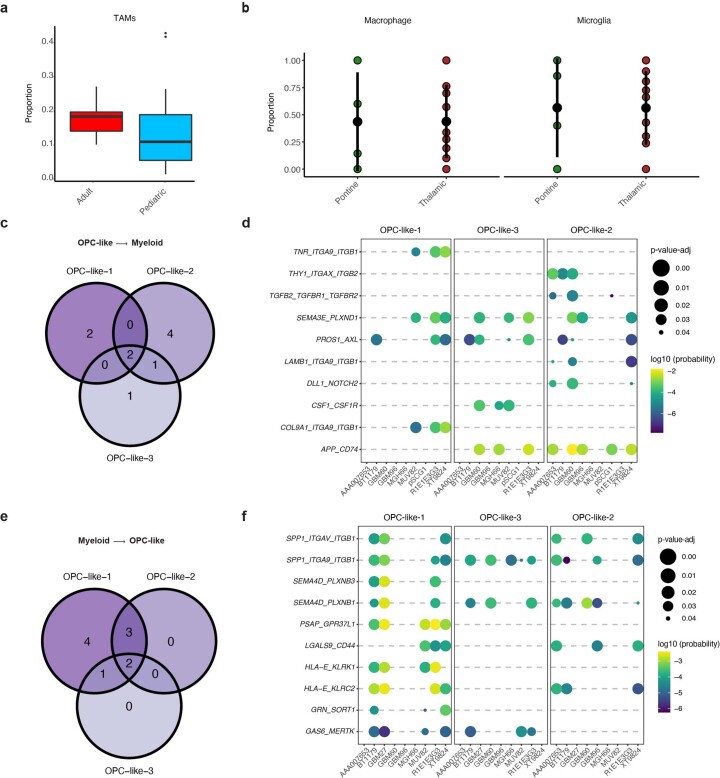
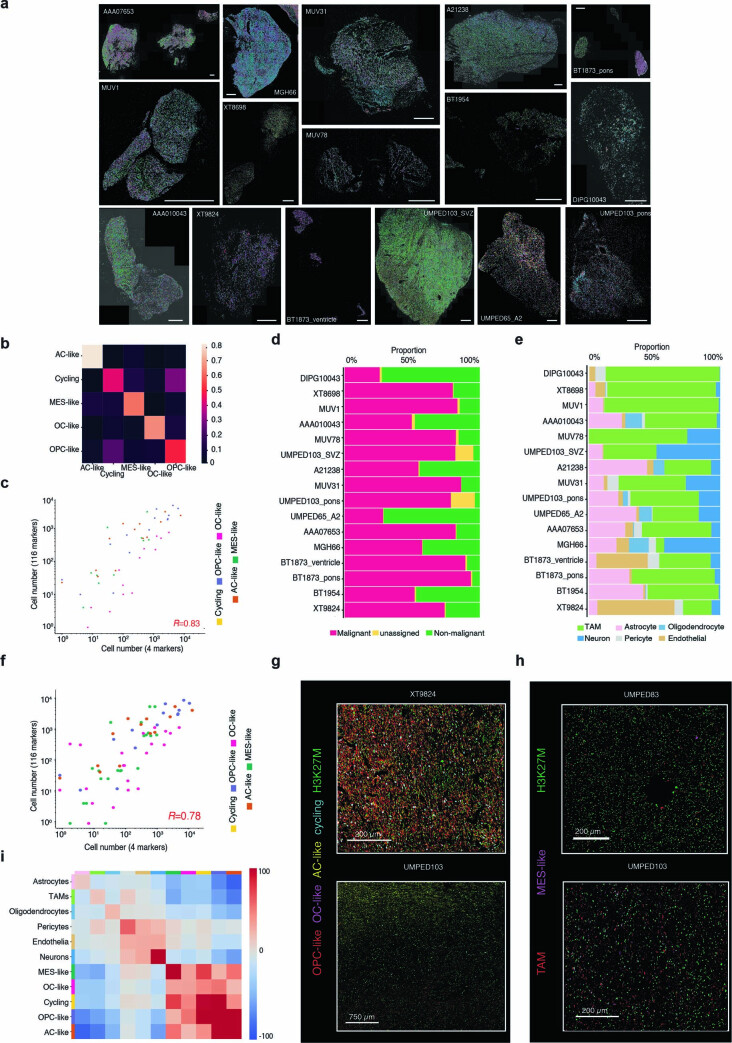
Histone 3 lysine27-to-methionine (H3-K27M) mutations most frequently occur in diffuse midline gliomas (DMGs) of the childhood pons but are also increasingly recognized in adults. Their potential heterogeneity at different ages and midline locations is vastly understudied. Here, through dissecting the single-cell transcriptomic, epigenomic and spatial architectures of a comprehensive cohort of patient H3-K27M DMGs, we delineate how age and anatomical location shape glioma cell-intrinsic and -extrinsic features in light of the shared driver mutation. We show that stem-like oligodendroglial precursor-like cells, present across all clinico-anatomical groups, display varying levels of maturation dependent on location. We reveal a previously underappreciated relationship between mesenchymal cancer cell states and age, linked to age-dependent differences in the immune microenvironment. Further, we resolve the spatial organization of H3-K27M DMG cell populations and identify a mitotic oligodendroglial-lineage niche. Collectively, our study provides a powerful framework for rational modeling and therapeutic interventions.
© 2022. The Author(s).
Conflict of interest statement
M.G.F. is a consultant for Twentyeight-Seven Therapeutics and Blueprint Medicines. M.N. is Scientific Advisor for 10X Genomics. M.M. is a SAB member for Cygnal Therapeutics. M.L. Suvà is an equity holder, scientific cofounder and advisory board member of Immunitas Therapeutics. K.L.L. is the founder and equity holder of Travera and receives consulting fees from BMS, Integragen, Rarecyte and research support from Lilly, BMS and Amgen. J.S. is now (but not when contributing to this manuscript) an employee of 10X Genomics. The remaining authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
