

K27M in canonical and noncanonical H3 variants occurs in distinct oligodendroglial cell lineages in brain midline gliomas
- PMID: 36471070
- PMCID: PMC9742294
- DOI: 10.1038/s41588-022-01205-w
K27M in canonical and noncanonical H3 variants occurs in distinct oligodendroglial cell lineages in brain midline gliomas
Abstract
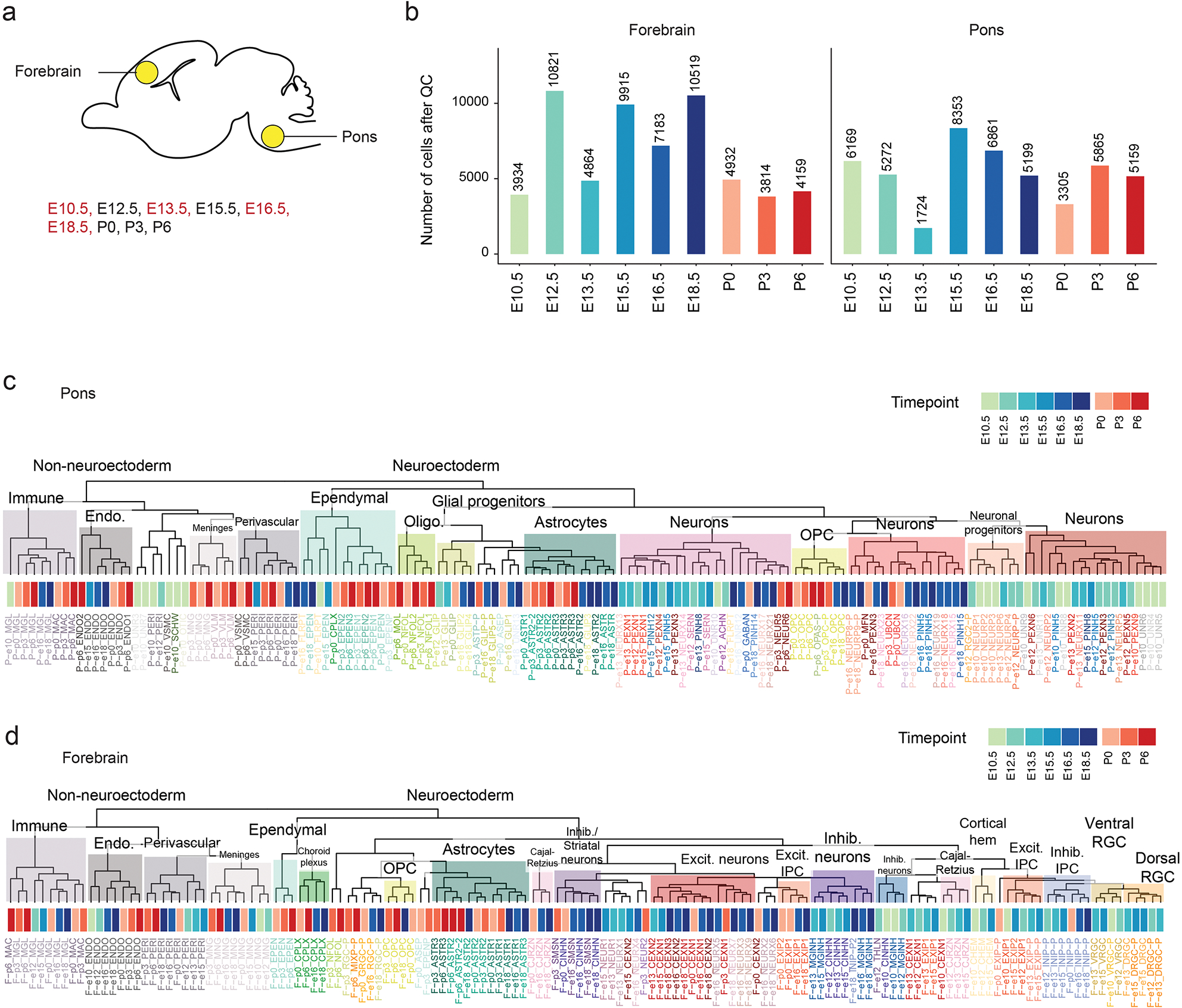
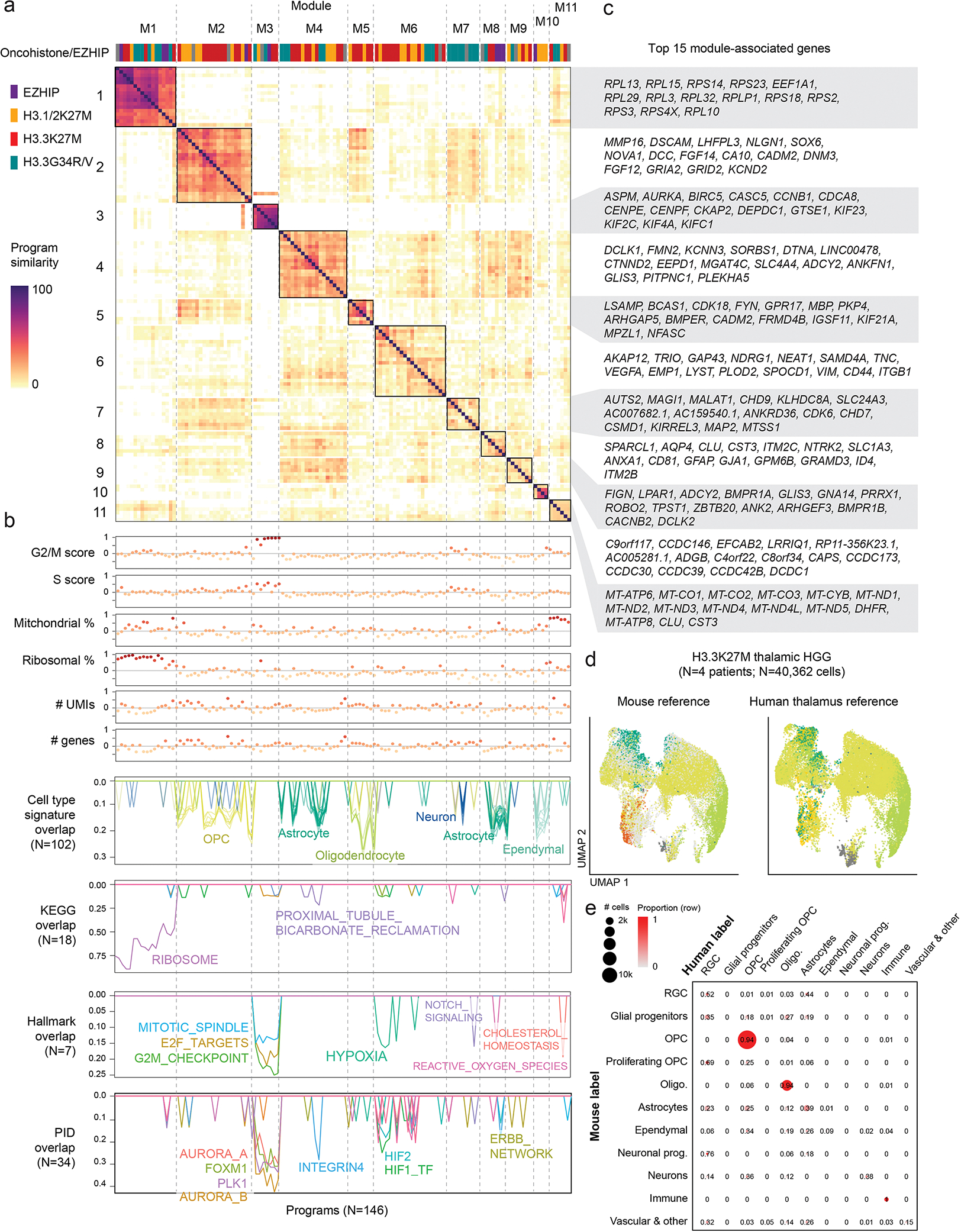
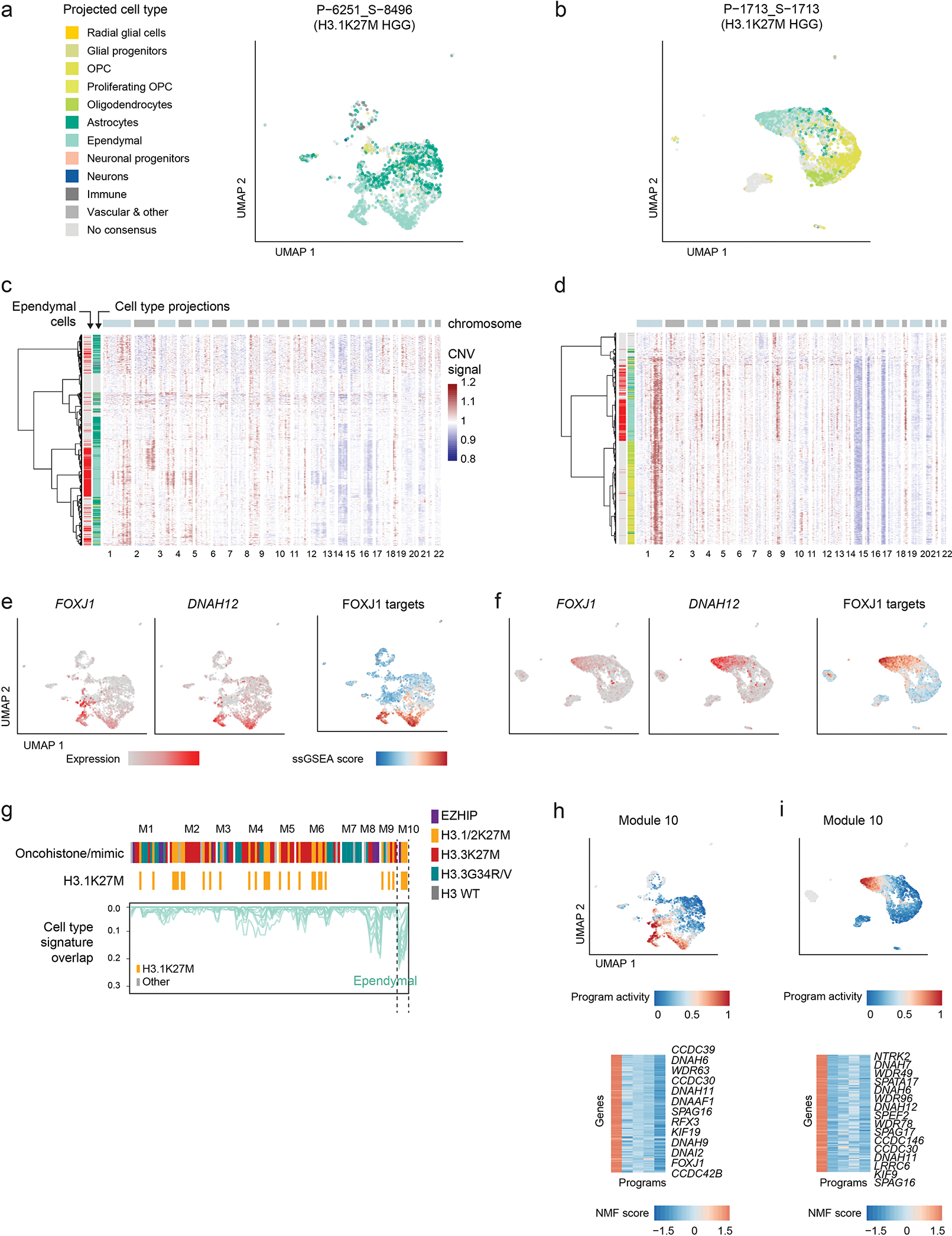
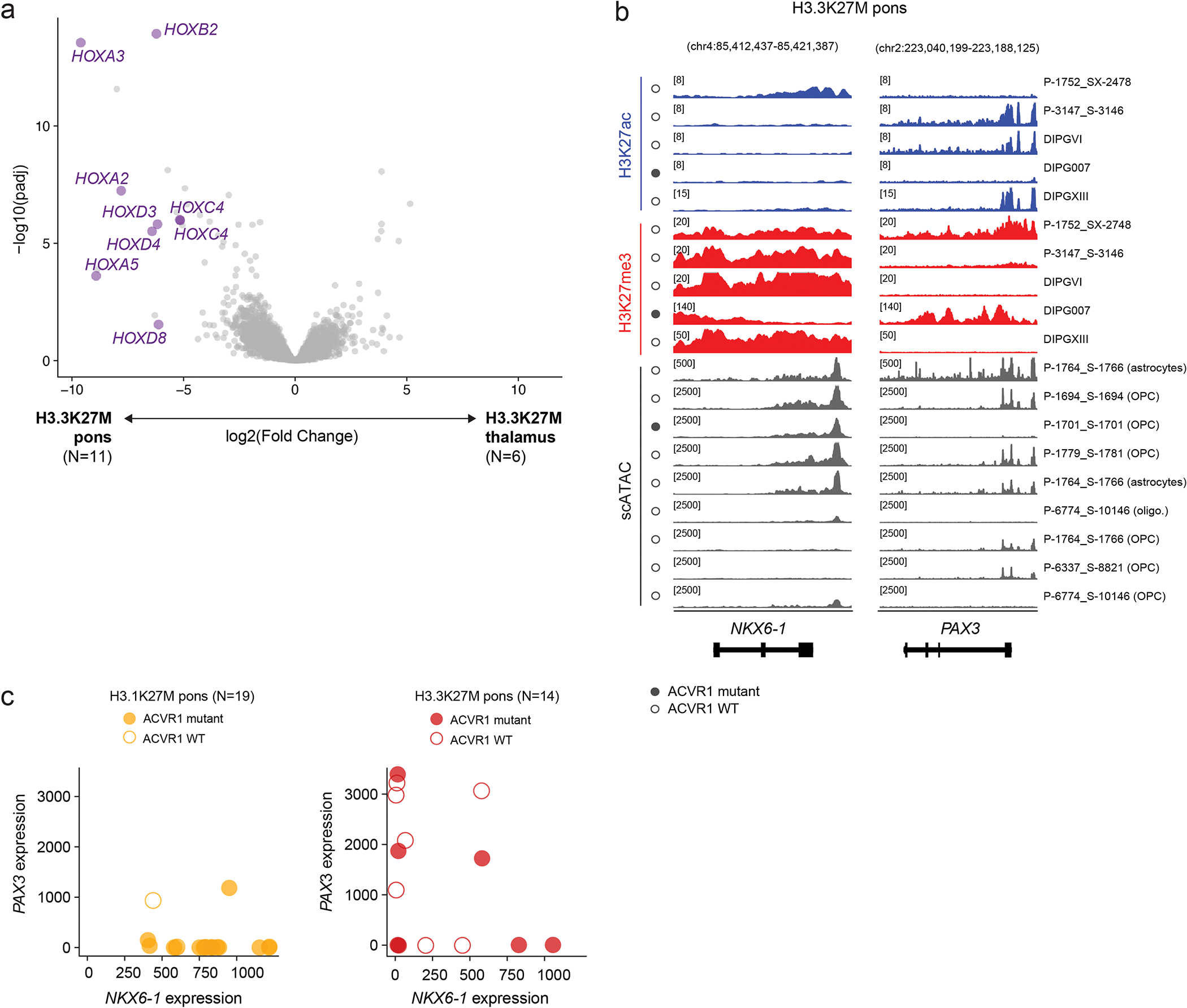
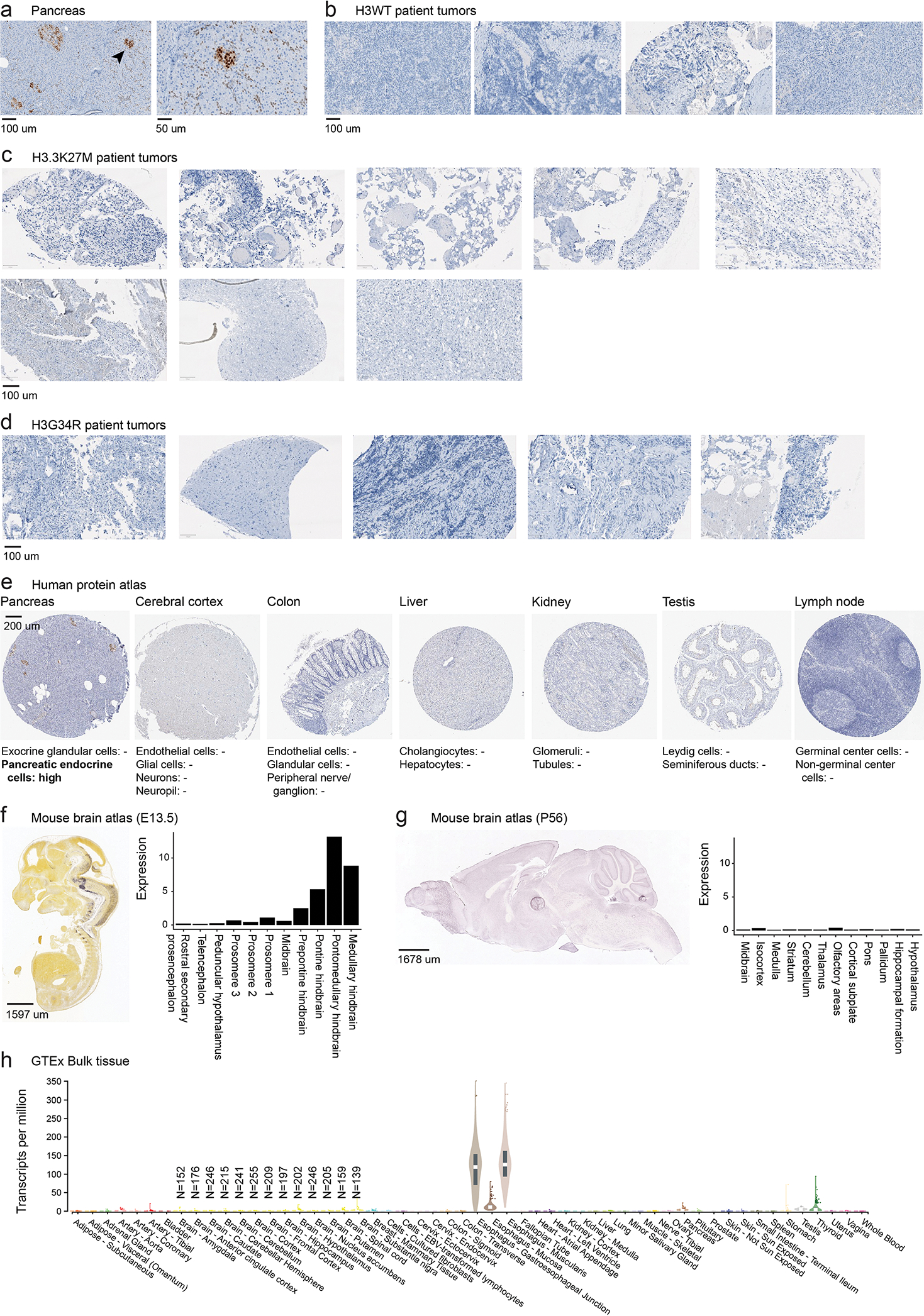
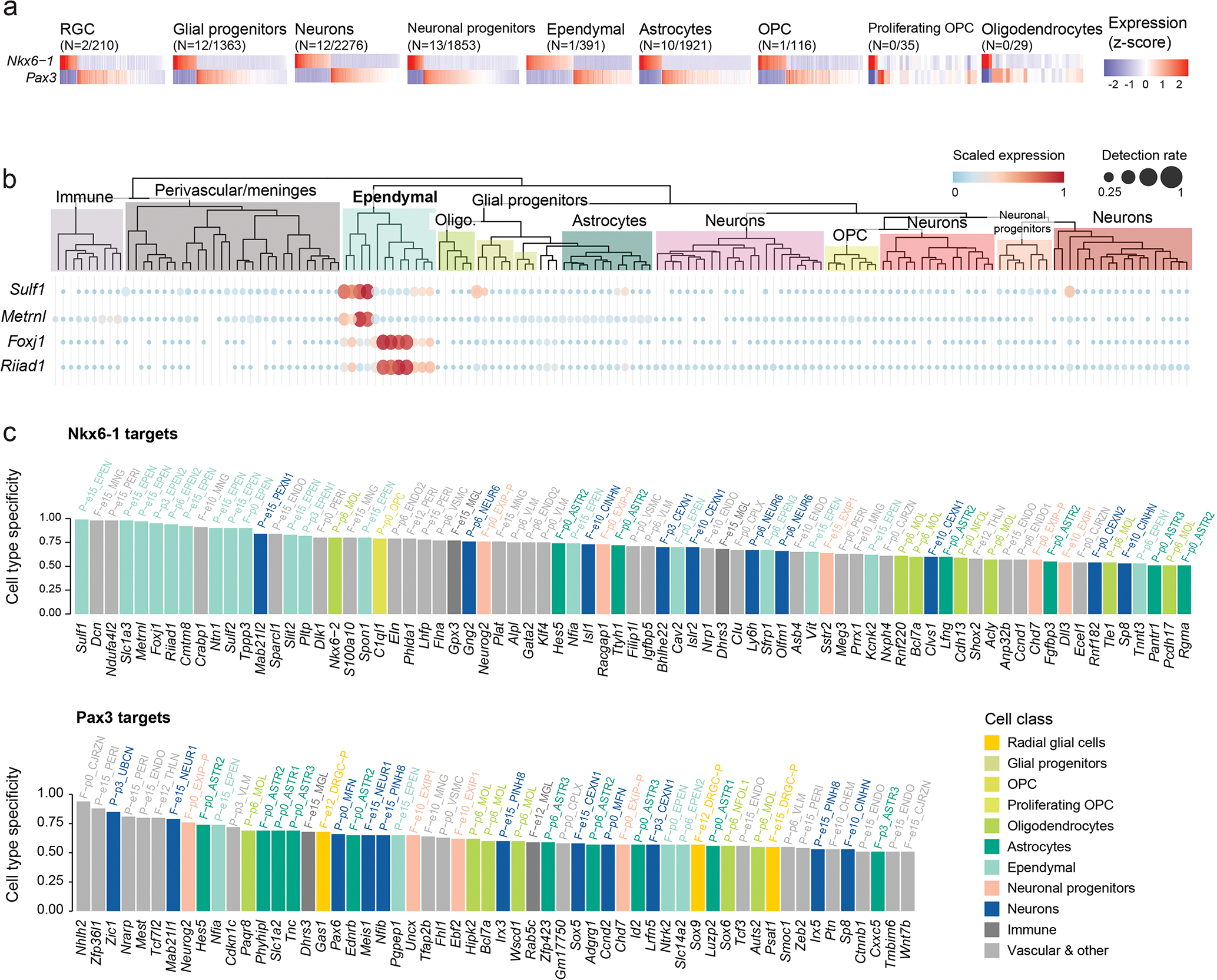
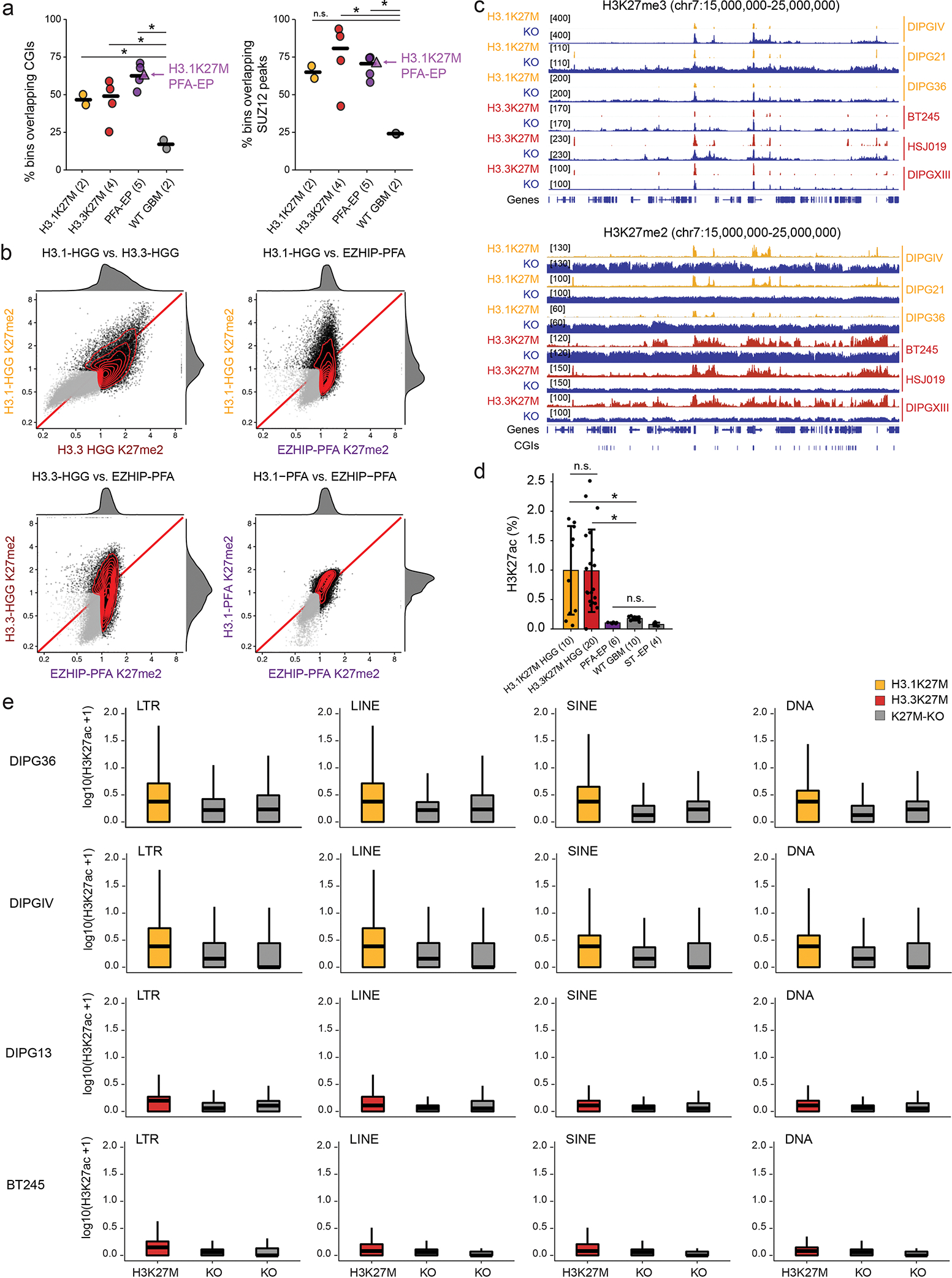
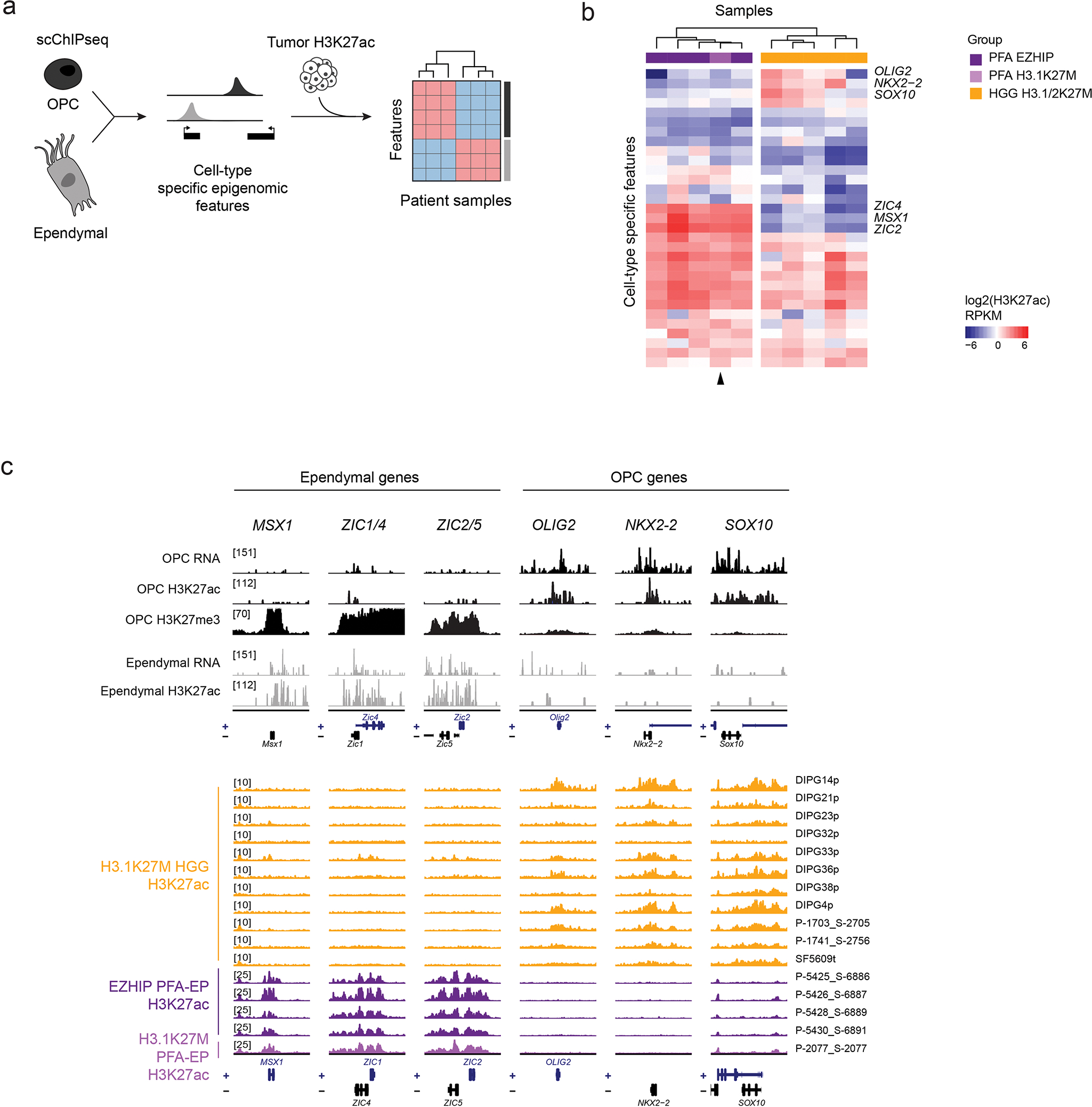
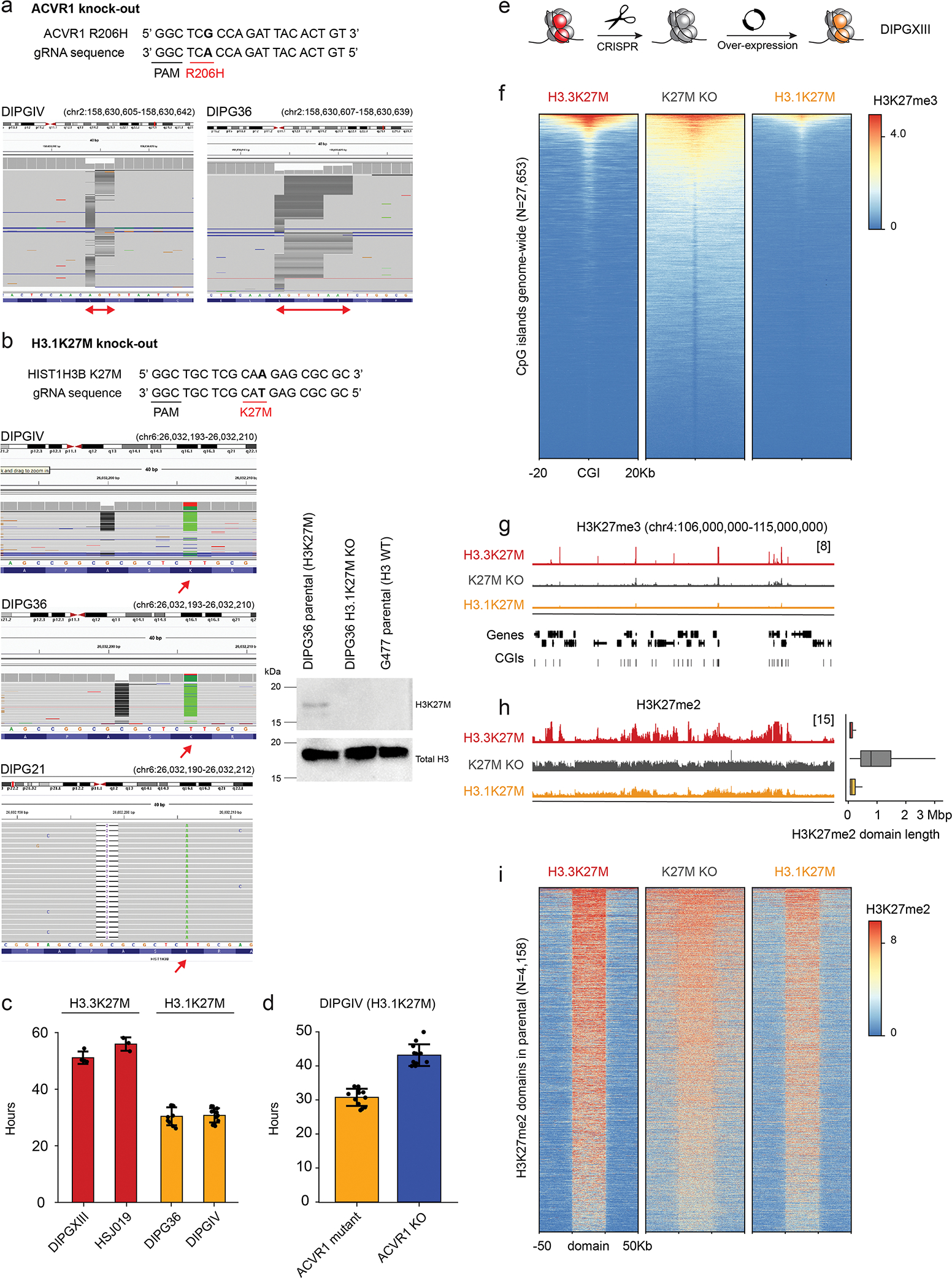
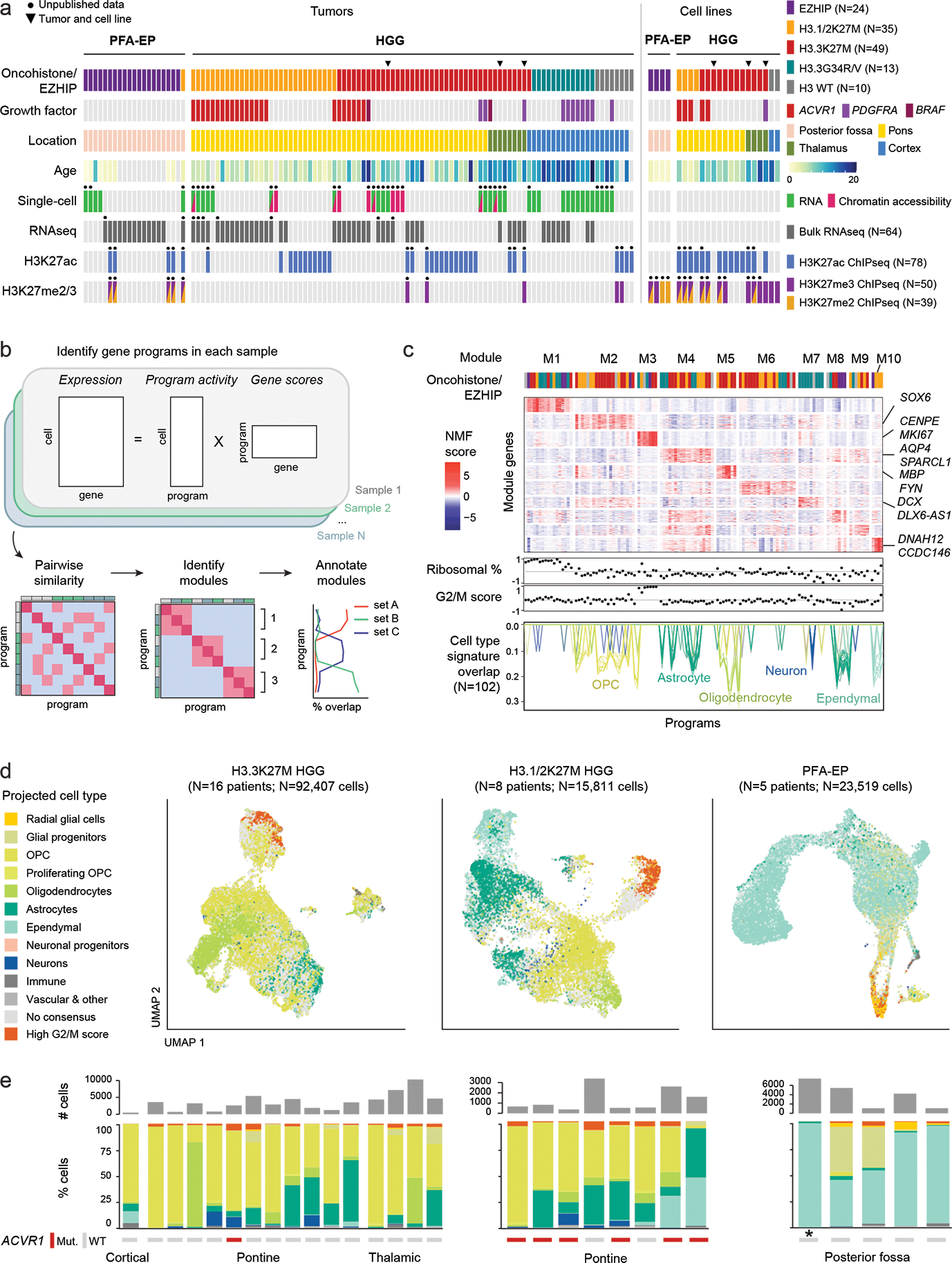
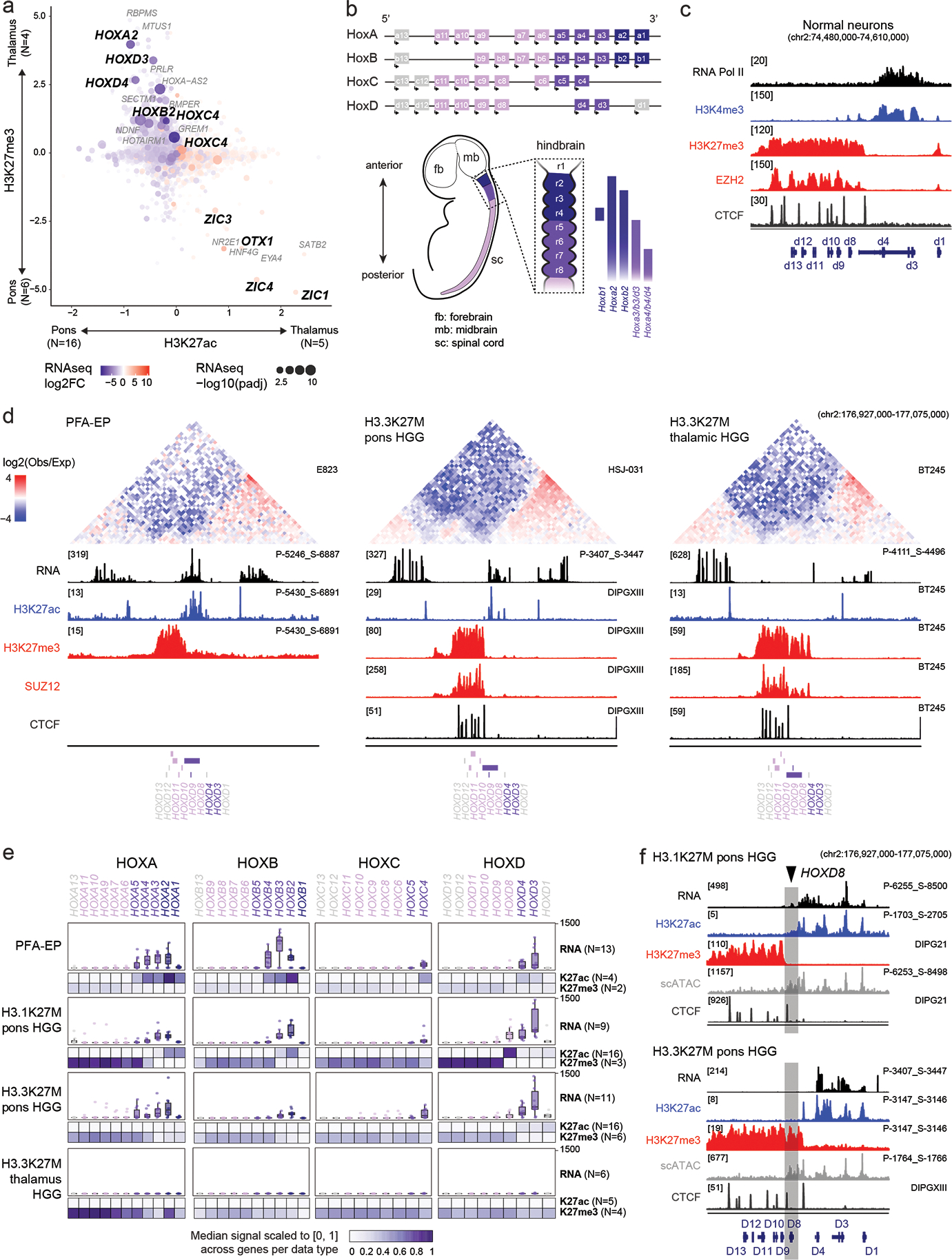
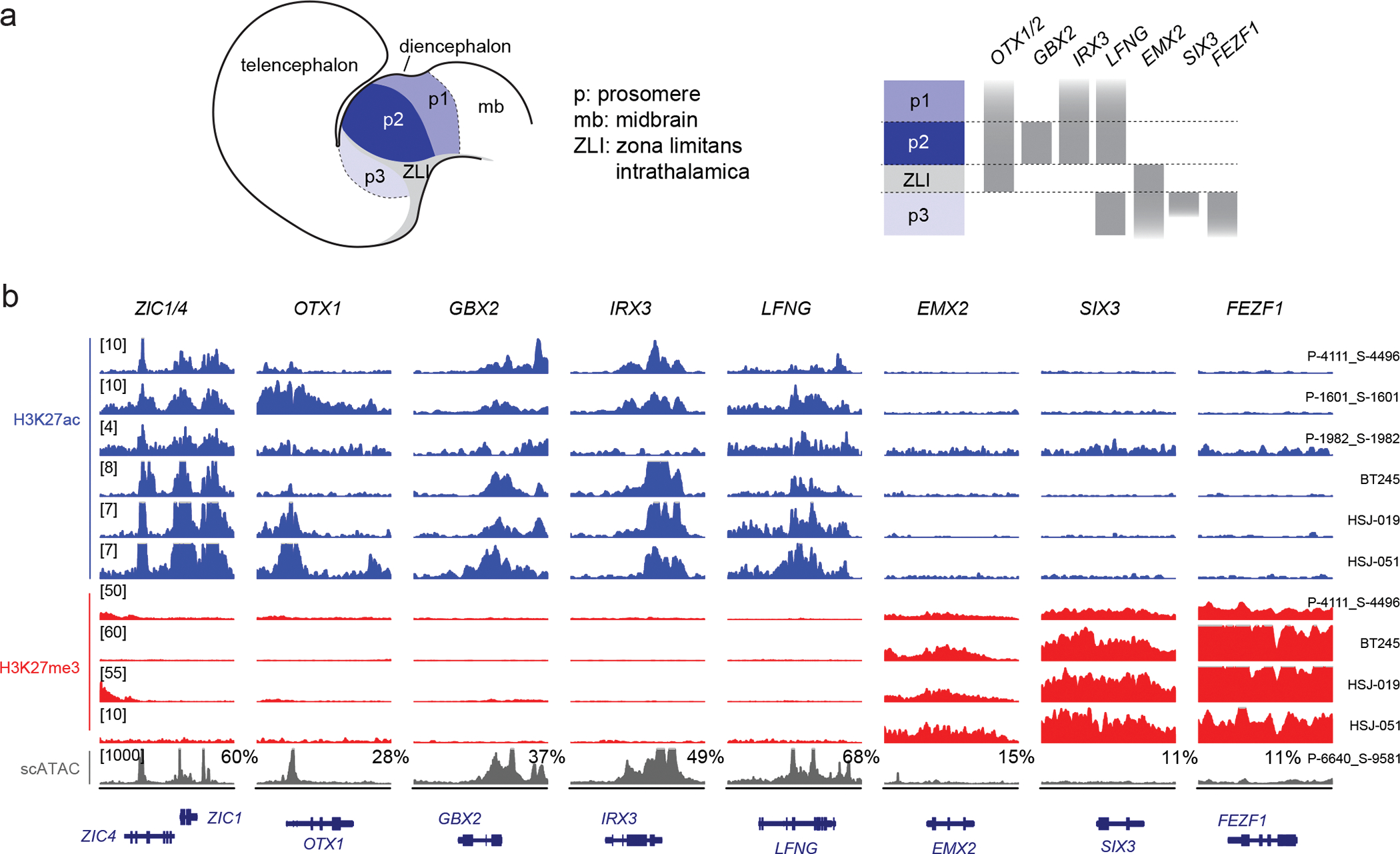
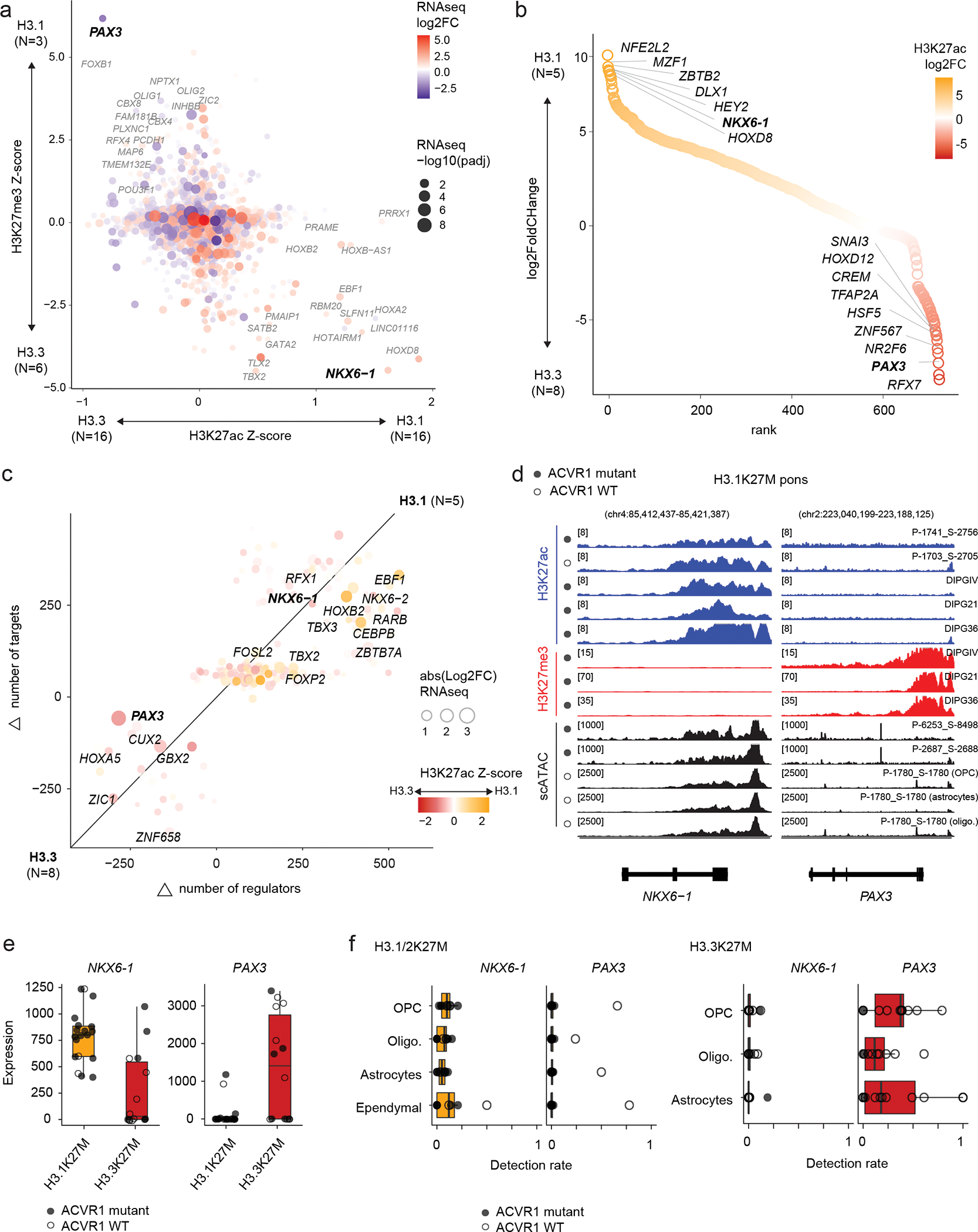
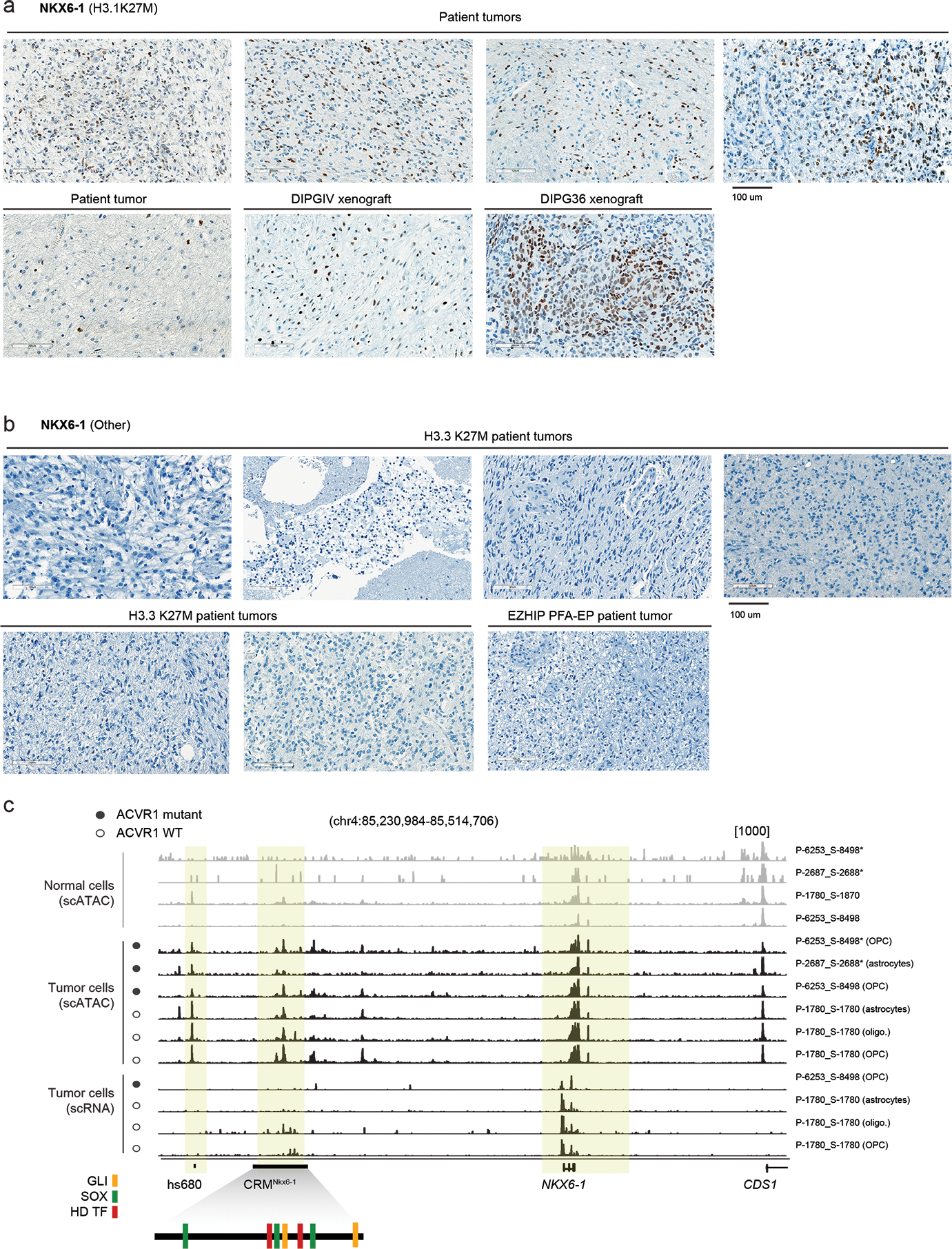
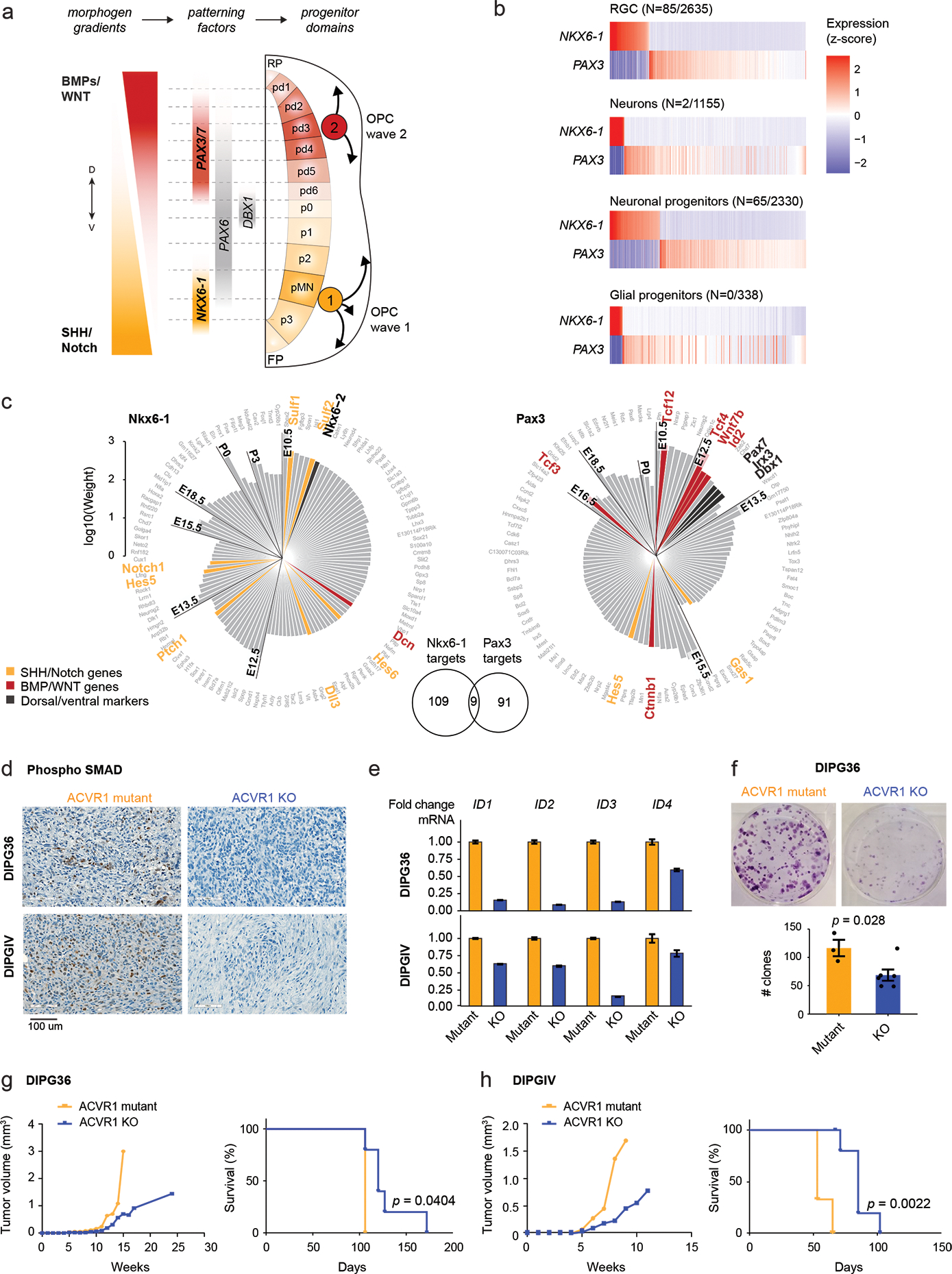
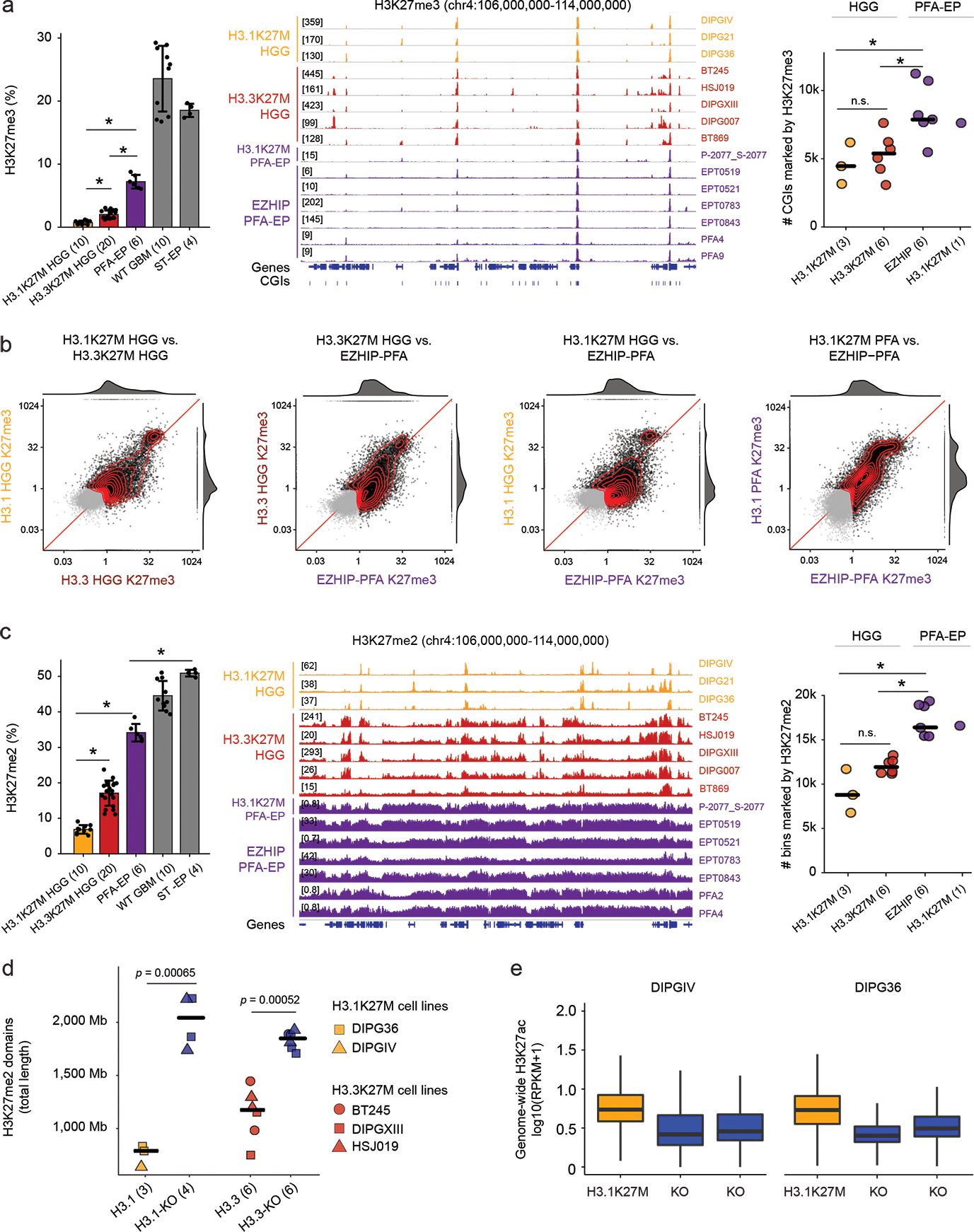
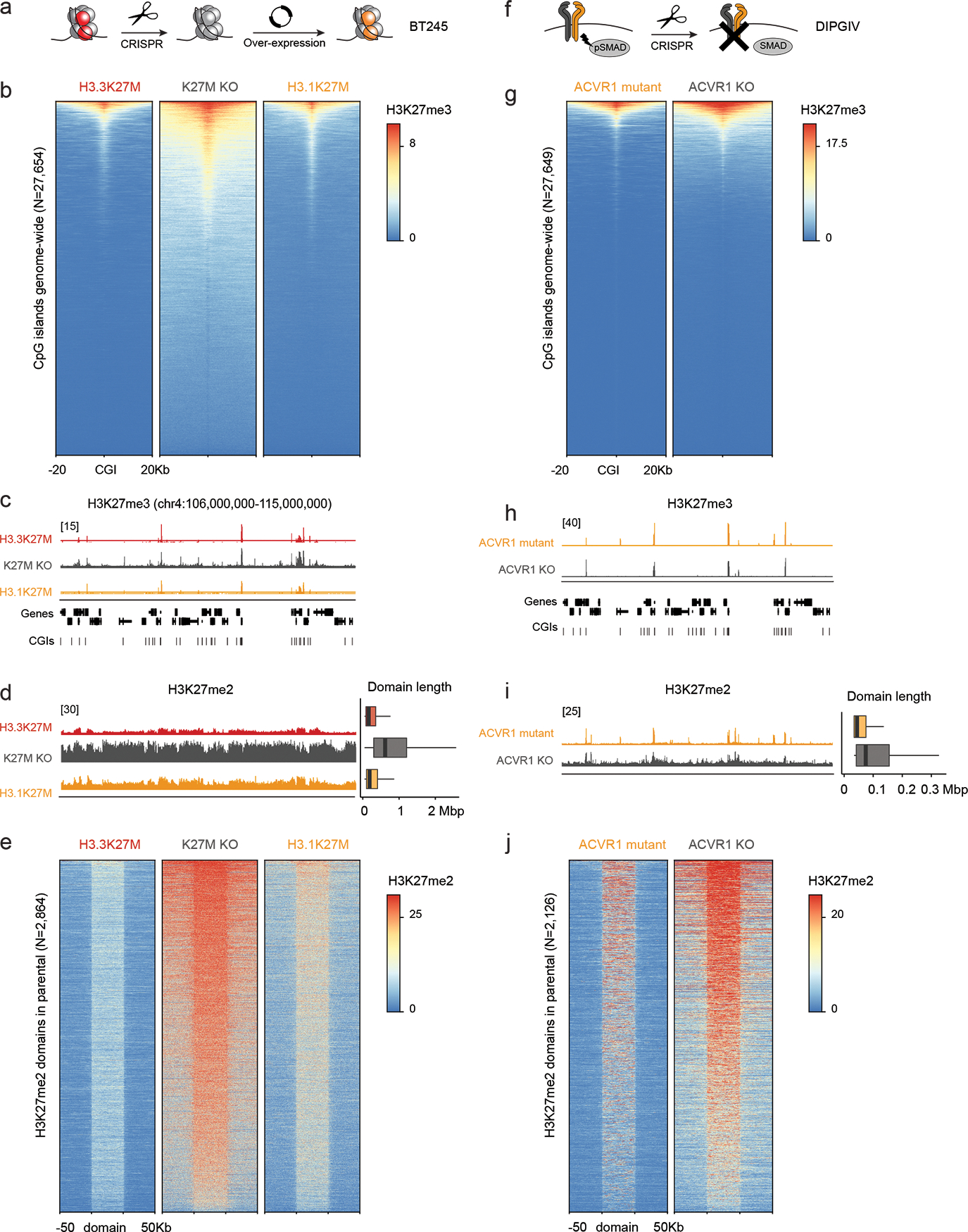
Canonical (H3.1/H3.2) and noncanonical (H3.3) histone 3 K27M-mutant gliomas have unique spatiotemporal distributions, partner alterations and molecular profiles. The contribution of the cell of origin to these differences has been challenging to uncouple from the oncogenic reprogramming induced by the mutation. Here, we perform an integrated analysis of 116 tumors, including single-cell transcriptome and chromatin accessibility, 3D chromatin architecture and epigenomic profiles, and show that K27M-mutant gliomas faithfully maintain chromatin configuration at developmental genes consistent with anatomically distinct oligodendrocyte precursor cells (OPCs). H3.3K27M thalamic gliomas map to prosomere 2-derived lineages. In turn, H3.1K27M ACVR1-mutant pontine gliomas uniformly mirror early ventral NKX6-1+/SHH-dependent brainstem OPCs, whereas H3.3K27M gliomas frequently resemble dorsal PAX3+/BMP-dependent progenitors. Our data suggest a context-specific vulnerability in H3.1K27M-mutant SHH-dependent ventral OPCs, which rely on acquisition of ACVR1 mutations to drive aberrant BMP signaling required for oncogenesis. The unifying action of K27M mutations is to restrict H3K27me3 at PRC2 landing sites, whereas other epigenetic changes are mainly contingent on the cell of origin chromatin state and cycling rate.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing Interests Statement
S.P. is a member of the advisory board for Bayer, Novartis and AstraZeneca and has received speaker fees from Bayer and Esai, outside of the submitted work. All other authors declare no competing interests.
Figures
References
-
- Schwartzentruber J et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 (2012). - PubMed
Methods-only references
-
- McInnes L & Healy J UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv [stat.ML] (2018).
-
- Waltman L & van Eck NJ A smart local moving algorithm for large-scale modularity-based community detection. The European Physical Journal B 86, 71 (2013).
-
- Nagy C et al. Single-nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat. Neurosci. 23, 771–781 (2020). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
