

Enhancer-promoter interactions and transcription are largely maintained upon acute loss of CTCF, cohesin, WAPL or YY1
- PMID: 36471071
- PMCID: PMC9729117
- DOI: 10.1038/s41588-022-01223-8
Enhancer-promoter interactions and transcription are largely maintained upon acute loss of CTCF, cohesin, WAPL or YY1
Abstract
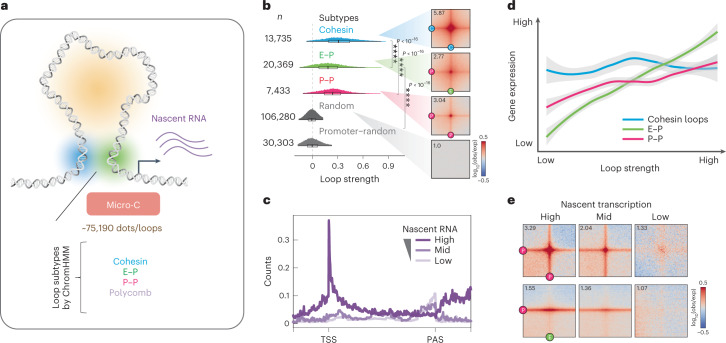
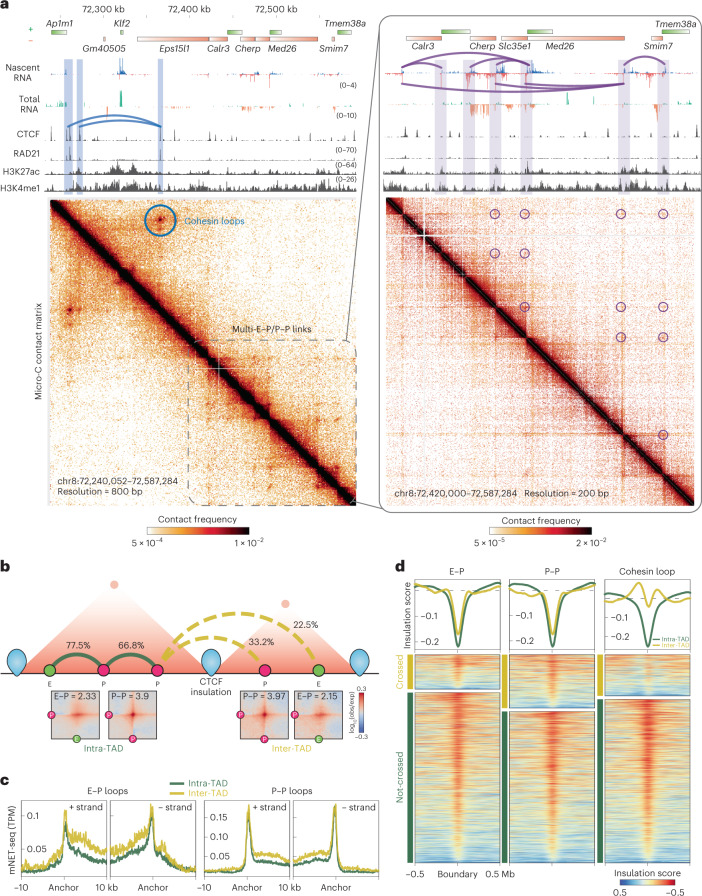
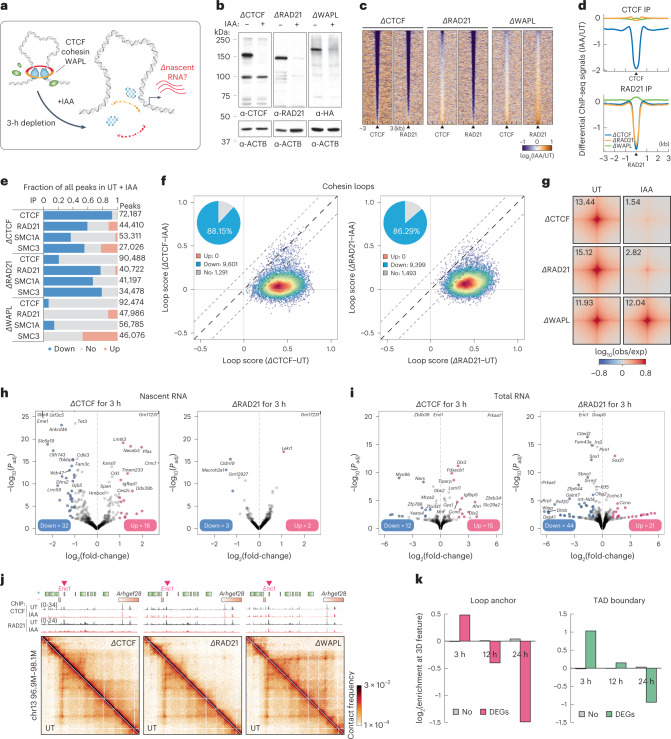
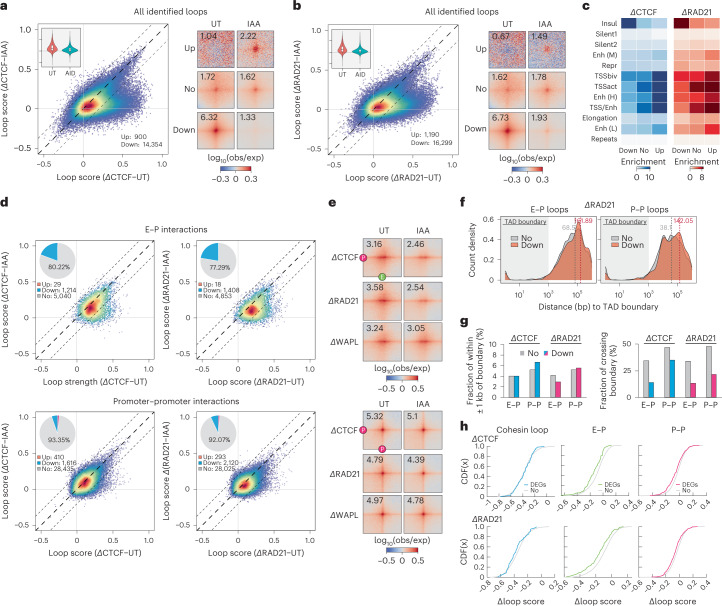
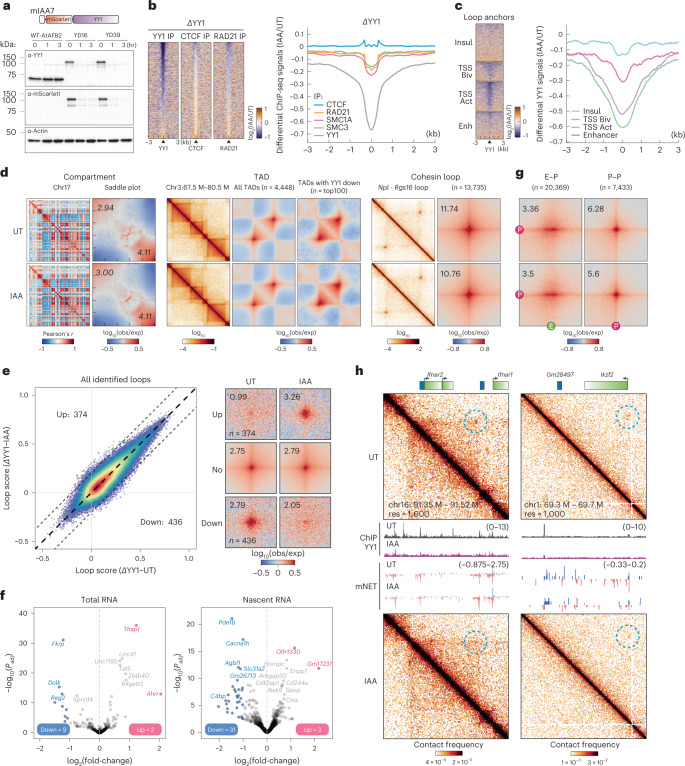
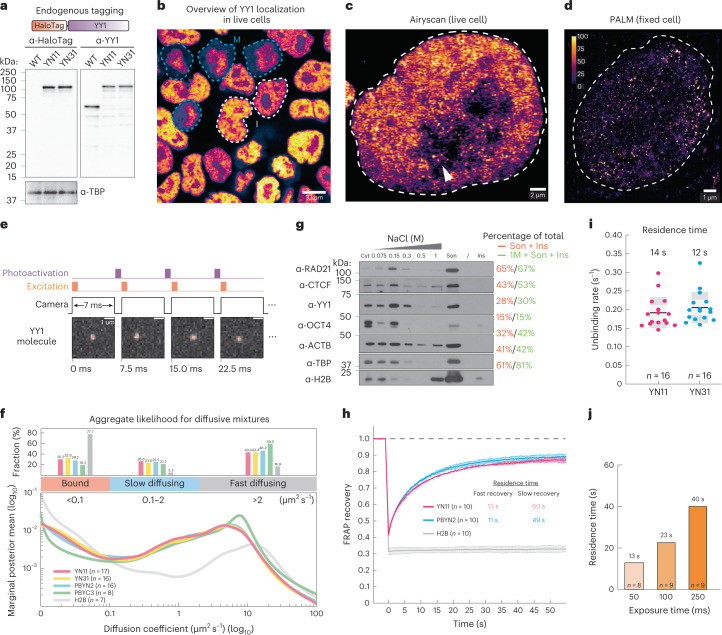
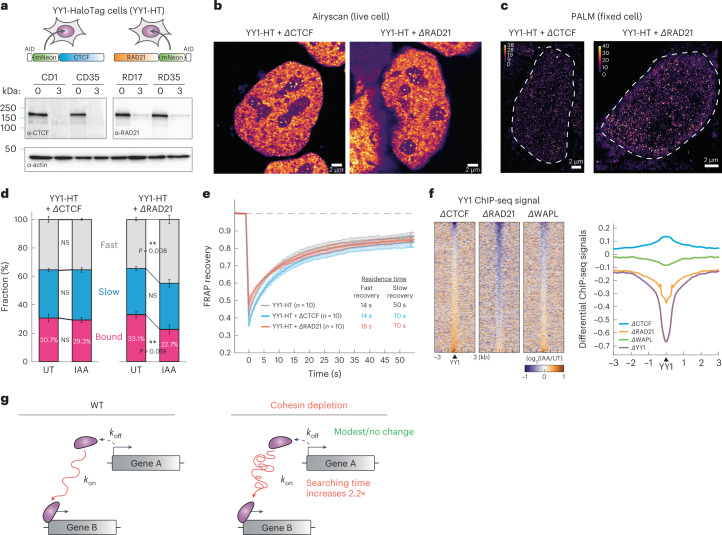
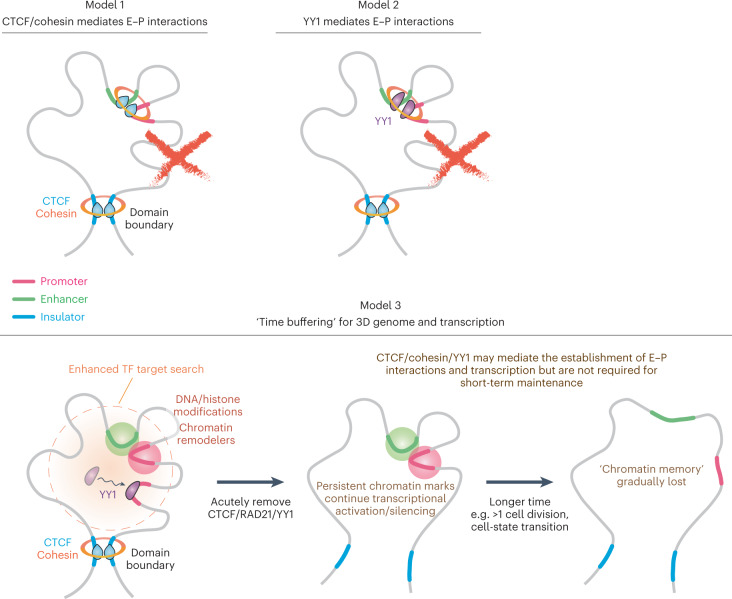
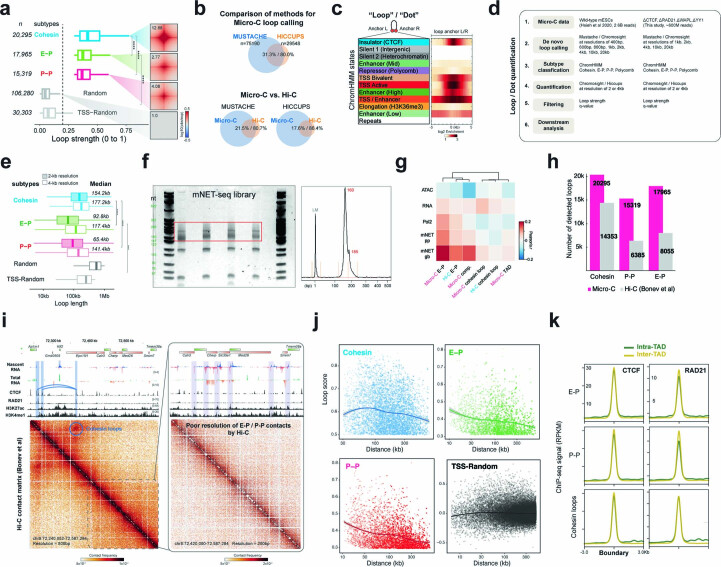
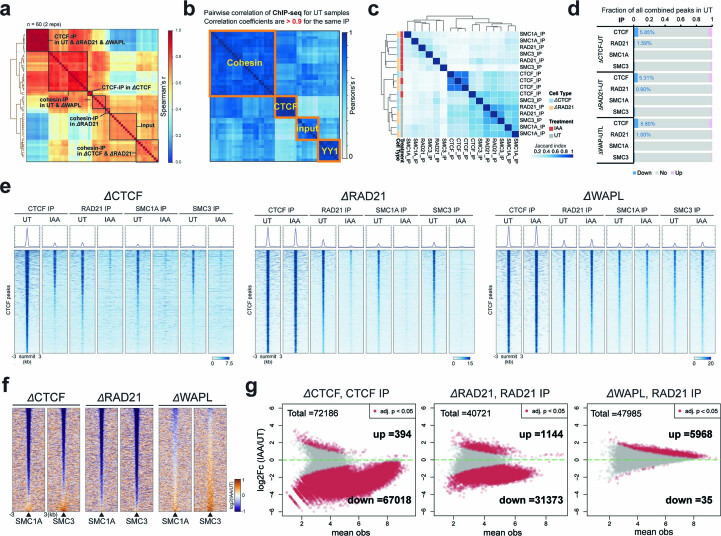
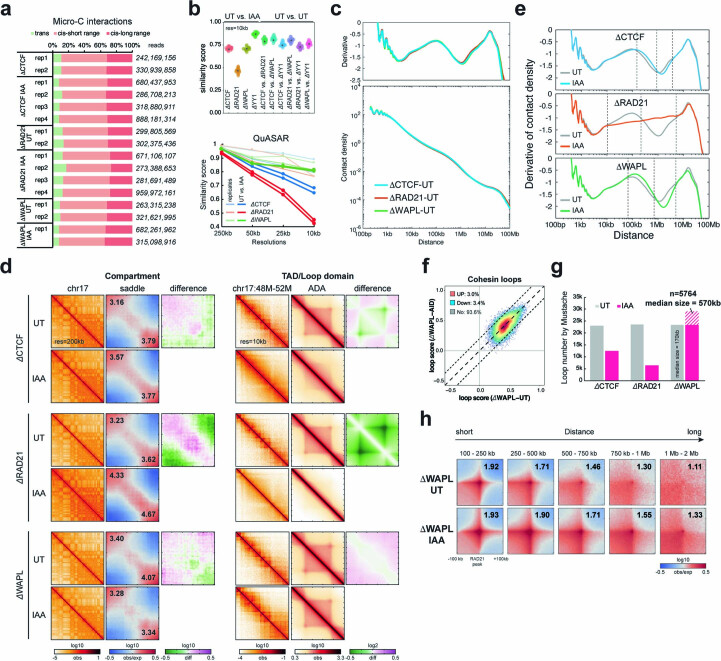
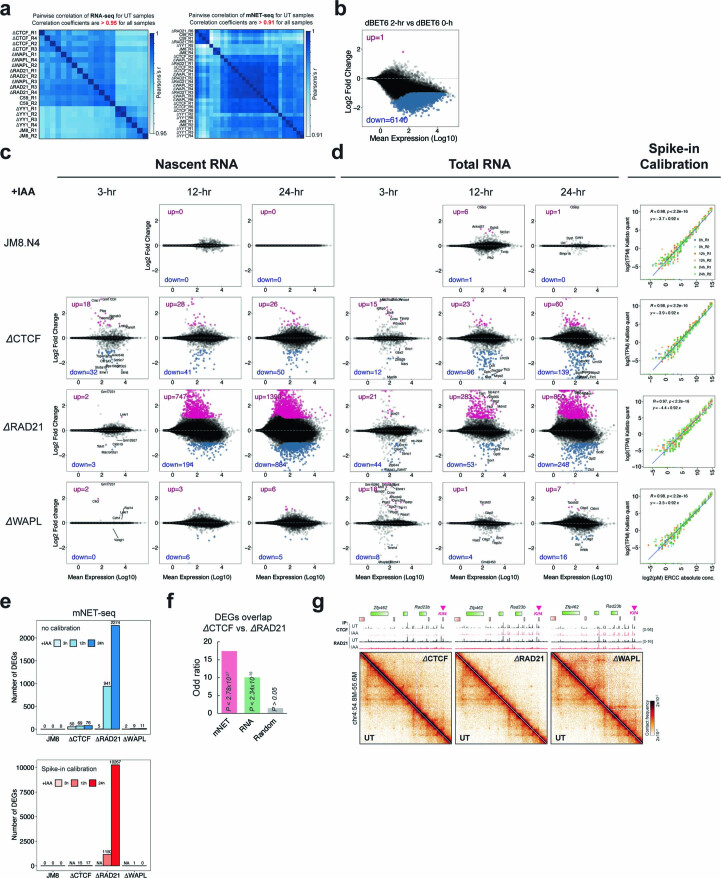
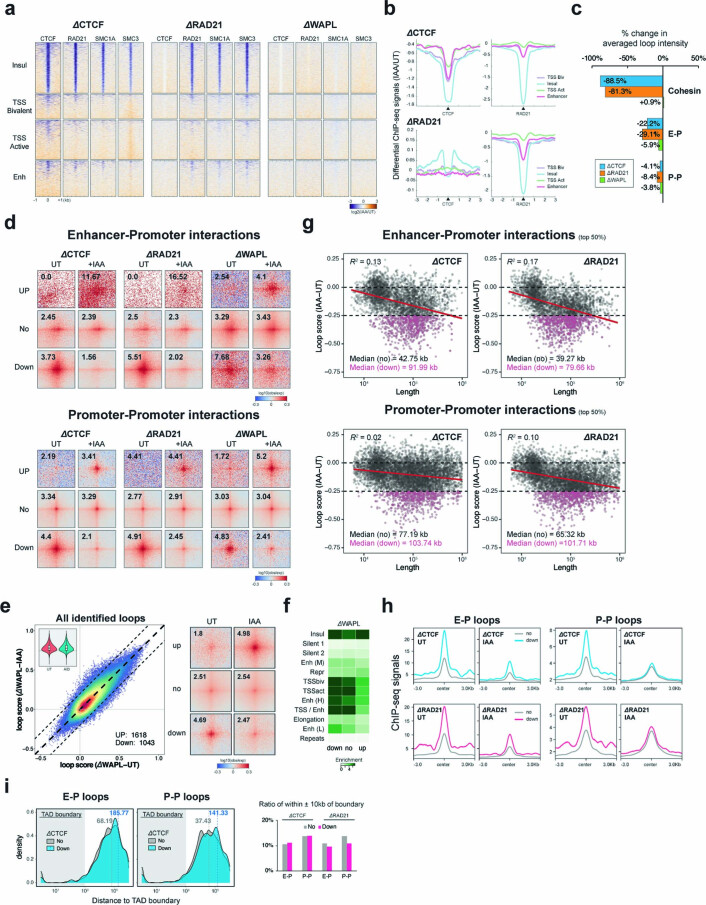
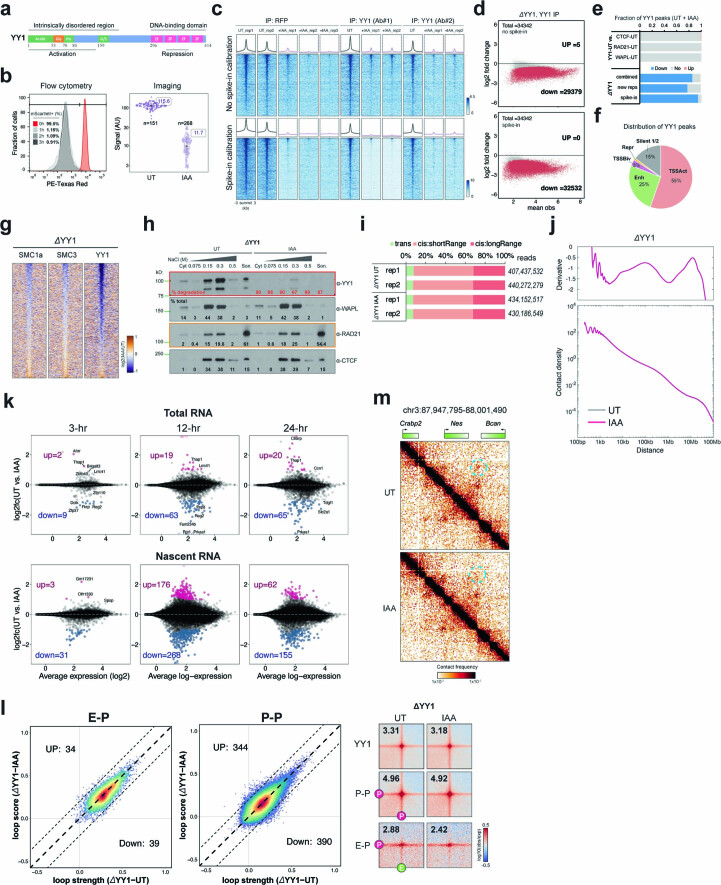
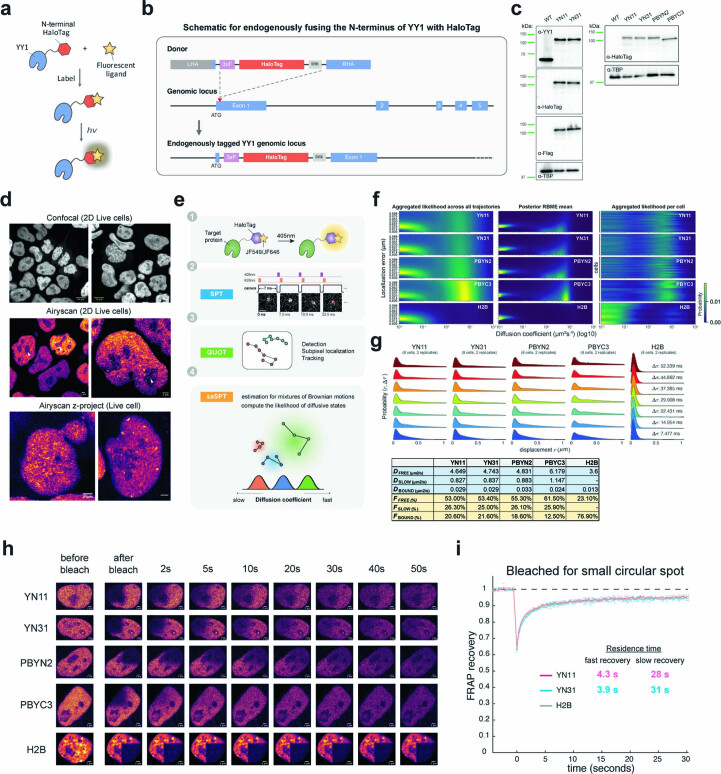
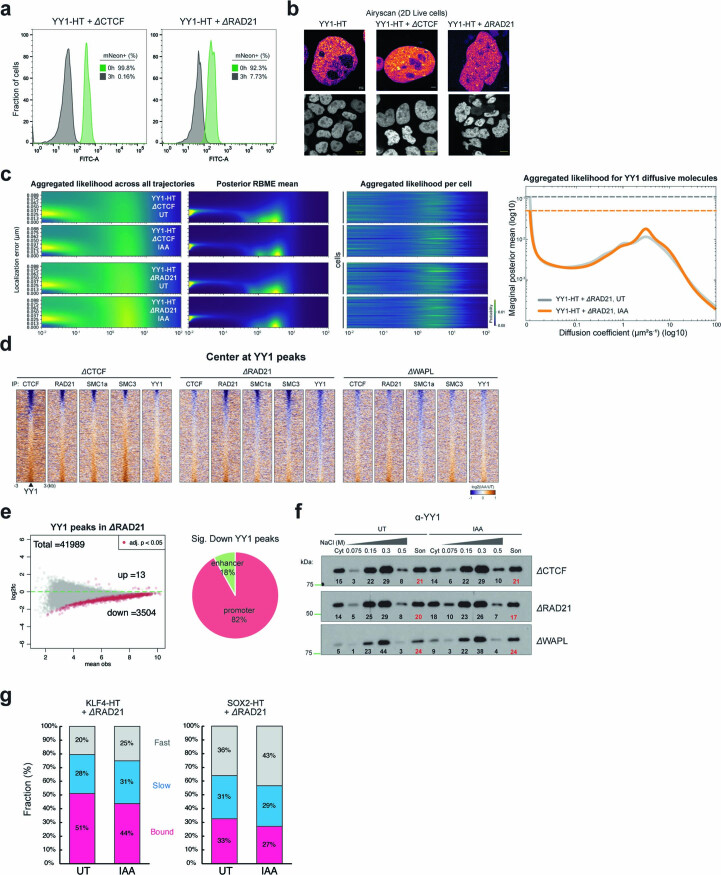
It remains unclear why acute depletion of CTCF (CCCTC-binding factor) and cohesin only marginally affects expression of most genes despite substantially perturbing three-dimensional (3D) genome folding at the level of domains and structural loops. To address this conundrum, we used high-resolution Micro-C and nascent transcript profiling in mouse embryonic stem cells. We find that enhancer-promoter (E-P) interactions are largely insensitive to acute (3-h) depletion of CTCF, cohesin or WAPL. YY1 has been proposed as a structural regulator of E-P loops, but acute YY1 depletion also had minimal effects on E-P loops, transcription and 3D genome folding. Strikingly, live-cell, single-molecule imaging revealed that cohesin depletion reduced transcription factor (TF) binding to chromatin. Thus, although CTCF, cohesin, WAPL or YY1 is not required for the short-term maintenance of most E-P interactions and gene expression, our results suggest that cohesin may facilitate TFs to search for and bind their targets more efficiently.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Jerkovic, I. & Cavalli, G. Understanding 3D genome organization by multidisciplinary methods. Nat. Rev. Mol. Cell Biol.10.1038/s41580-021-00362-w (2021). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
