

The mutation spectrum and ethnic distribution of non-hepatorenal tyrosinemia (types II, III)
- PMID: 36471409
- PMCID: PMC9724276
- DOI: 10.1186/s13023-022-02579-0
The mutation spectrum and ethnic distribution of non-hepatorenal tyrosinemia (types II, III)
Abstract
Background: Different types of non-hepatorenal tyrosinemia are among the rare forms of tyrosinemia. Tyrosinemia type II and III are autosomal recessive disorders caused by pathogenic variants in the tyrosine aminotransferase (TAT), and 4-hydroxyphenyl-pyruvate dioxygenas (HPPD) genes, respectively. There are still unclarified aspects in their clinical presentations, mutational spectrum, and genotype-phenotype correlation.
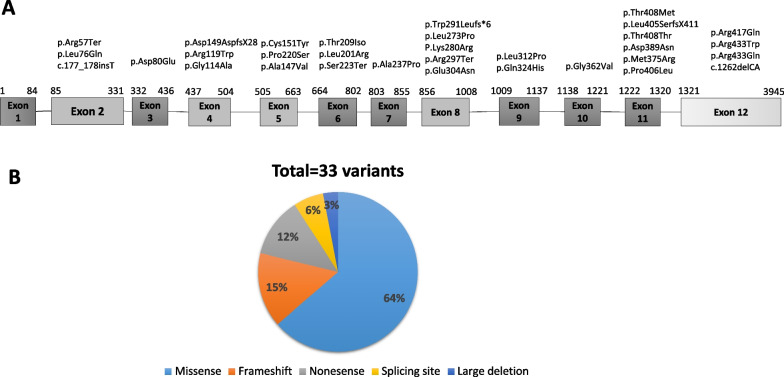
Main body: In this study, we evaluated the spectrum of TAT and HHPD gene mutations in patients with tyrosinemia type II and III. Moreover, biochemical and clinical findings are evaluated to establish a genotype-phenotype relationship in the above-mentioned patients. Thirty-three TAT variants have been reported in 42 families, consisting of 21 missense variants, 5 frameshift variants, 4 nonsense variants, 2 variants that primarily cause splicing site, and 1 skipping complete exon (large deletion). The most common variant is p.Arg57Ter, causing a splicing defect, and resulting in premature termination of translation, which was found in 10 patients from 3 families. In HPPD gene, eleven variants in 16 patients have been reported including 7 missense variants, 2 nonsense variants, 1 splice defect, and 1 frameshift variant so far. All variants are unique, except for p.Tyr160Cys, which is a missense variant found in two different patients. Regarding genotype-phenotype correlations, in 90% of tyrosinemia type II patients, positive clinical and biochemical correlations with a detected variant are observed. In HPPD gene, due to the small number of patients, it is not possible to make a definite conclusion.
Conclusion: This is the first large review of variants in TAT and HPPD, highlighting the wide spectrum of disease-causing mutations. Such information is beneficial for the establishment of a privileged mutation screening process in a specific region or ethnic group.
Keywords: 4-Hydroxyphenylpyruvate dioxygenase; Genotype; Mutations; Tyrosine aminotransferase; Tyrosinemia.
© 2022. The Author(s).
Conflict of interest statement
All authors declare no competing interests.
Figures
Similar articles
-
Mutations in the 4-hydroxyphenylpyruvate dioxygenase gene (HPD) in patients with tyrosinemia type III.Hum Genet. 2000 Jun;106(6):654-62. doi: 10.1007/s004390000307. Hum Genet. 2000. PMID: 10942115
-
Tyrosinemia type II: Mutation update, 11 novel mutations and description of 5 independent subjects with a novel founder mutation.Clin Genet. 2017 Sep;92(3):306-317. doi: 10.1111/cge.13003. Epub 2017 May 18. Clin Genet. 2017. PMID: 28255985
-
Mutation spectrum of Tyrosinemia type I in Iran, A retrospective cohort study.Eur J Med Genet. 2024 Oct;71:104970. doi: 10.1016/j.ejmg.2024.104970. Epub 2024 Sep 10. Eur J Med Genet. 2024. PMID: 39260601
-
Novel HPD mutation p.A244V compound with p.T219M causing tyrosinemia type III in a Chinese girl and review of the genotype-phenotype spectrum.Mol Genet Genomic Med. 2024 Jan;12(1):e2298. doi: 10.1002/mgg3.2298. Epub 2023 Oct 10. Mol Genet Genomic Med. 2024. PMID: 37817461 Free PMC article. Review.
-
The genetic tyrosinemias.Am J Med Genet C Semin Med Genet. 2006 May 15;142C(2):121-6. doi: 10.1002/ajmg.c.30092. Am J Med Genet C Semin Med Genet. 2006. PMID: 16602095 Review.
Cited by
-
The Establishment of Expanded Newborn Screening in Rural Areas of a Developing Country: A Model from Health Regions 7 and 8 in Thailand.Int J Neonatal Screen. 2025 Apr 12;11(2):26. doi: 10.3390/ijns11020026. Int J Neonatal Screen. 2025. PMID: 40265447 Free PMC article.
References
-
- Chakrapani A, Gissen P, McKiernan P. Disorders of tyrosine metabolism. In: Saudubray JM, van den Berghe G, Walter JH, editors. Inborn Metabolic diseases: diagnosis and treatment. Berlin: Springer; 2012. pp. 265–276.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
