

Carboxylic Acids as Adaptive Functional Groups in Metallaphotoredox Catalysis
- PMID: 36472093
- PMCID: PMC10680106
- DOI: 10.1021/acs.accounts.2c00607
Carboxylic Acids as Adaptive Functional Groups in Metallaphotoredox Catalysis
Abstract
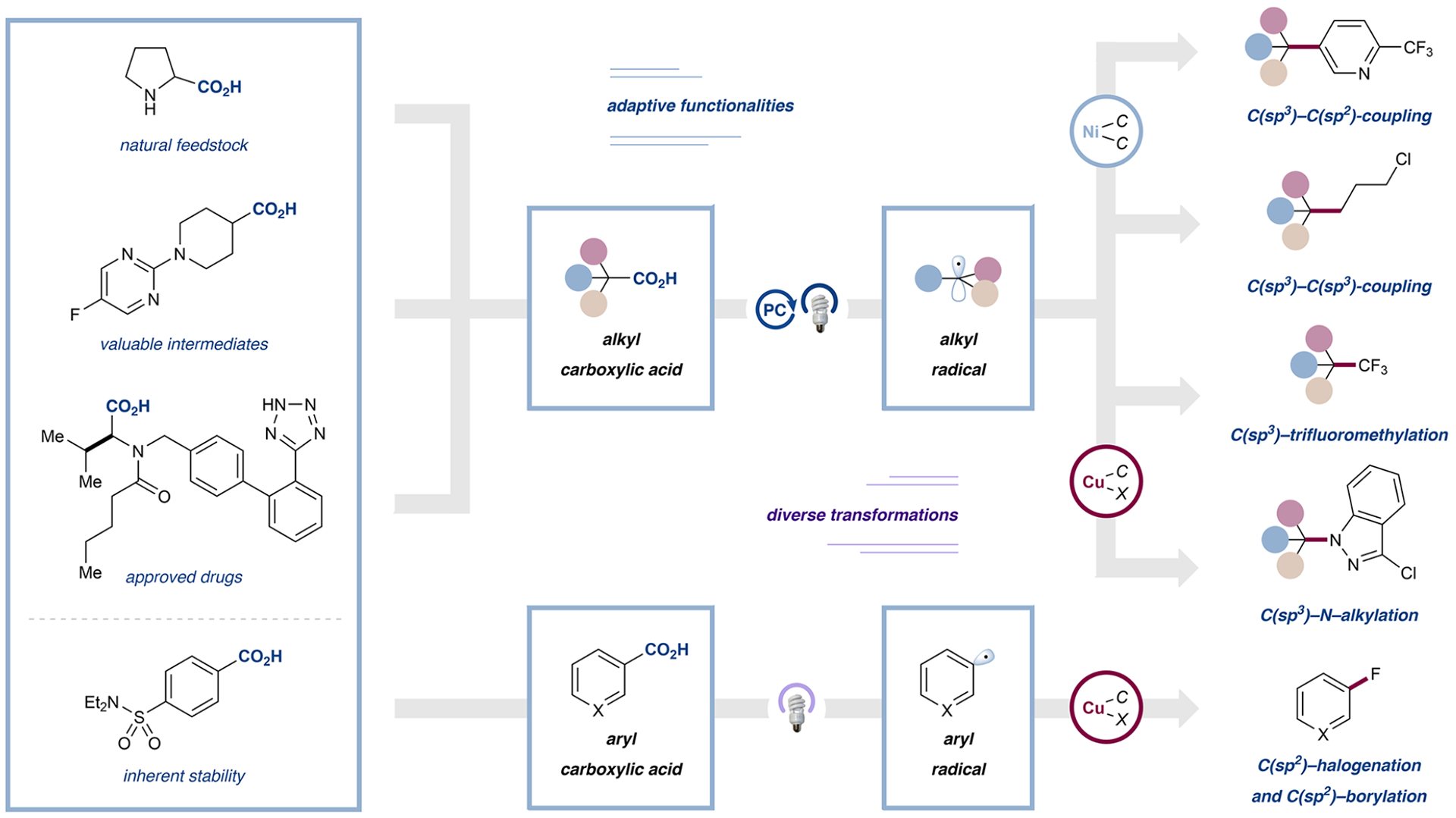
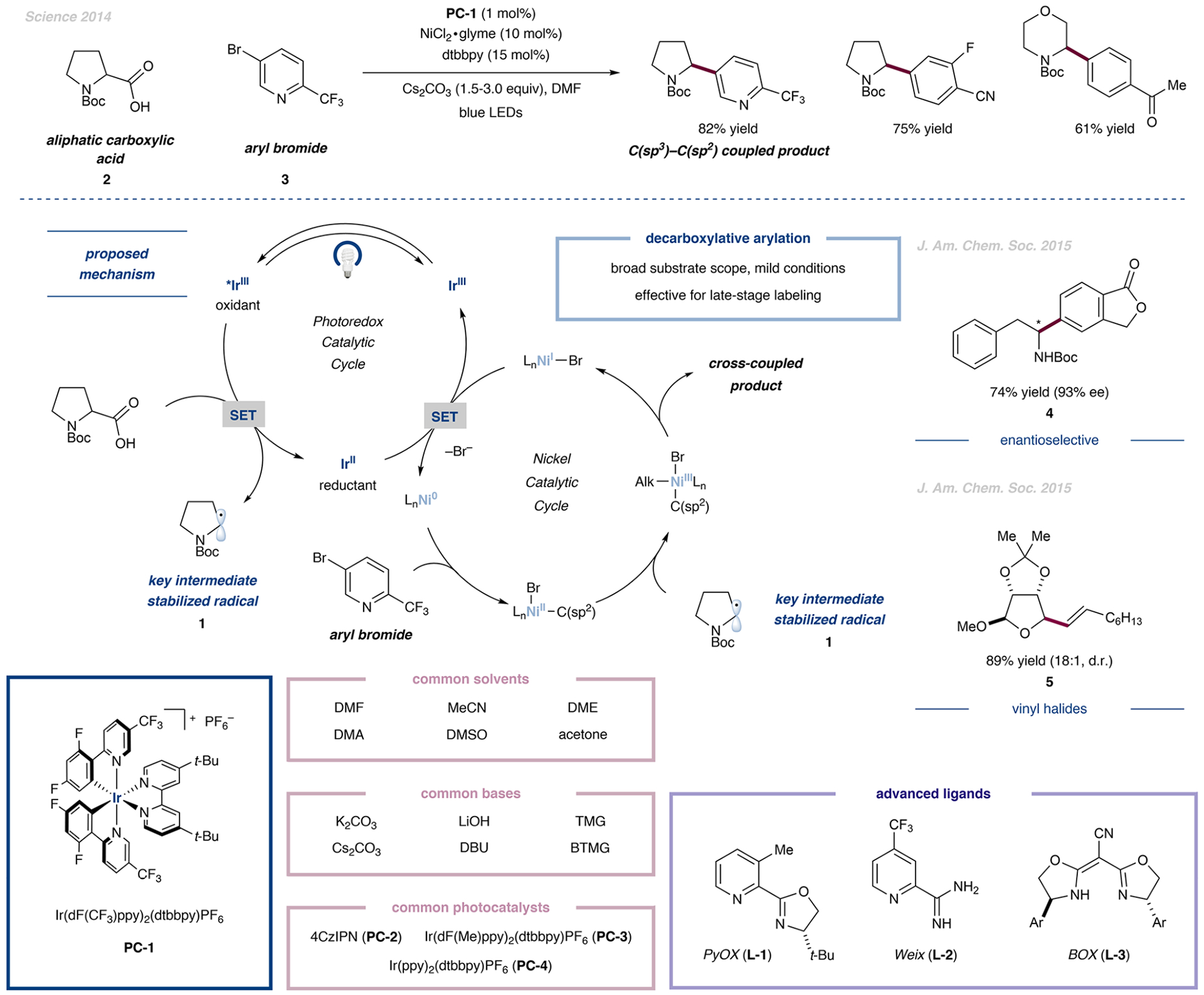
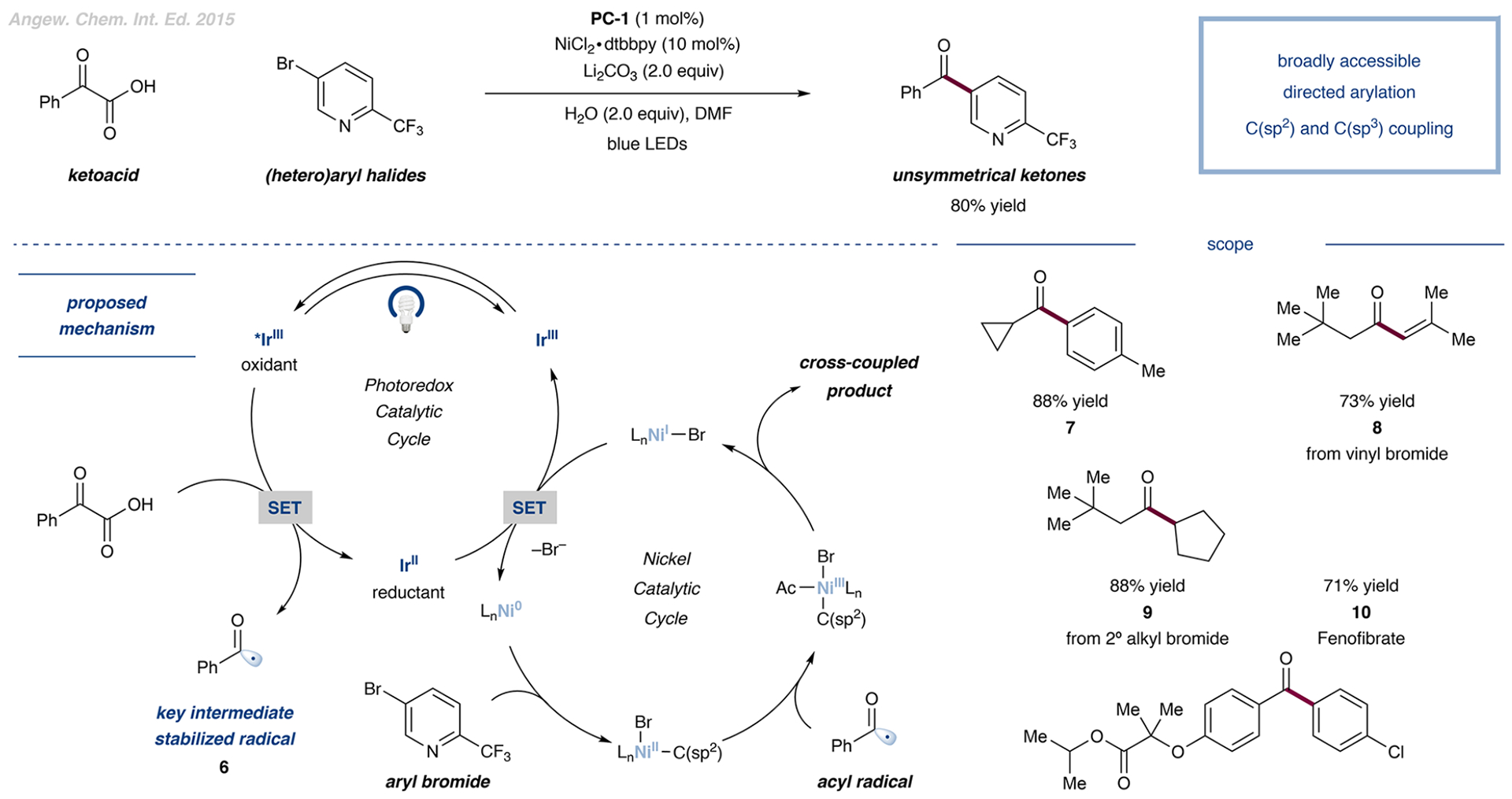
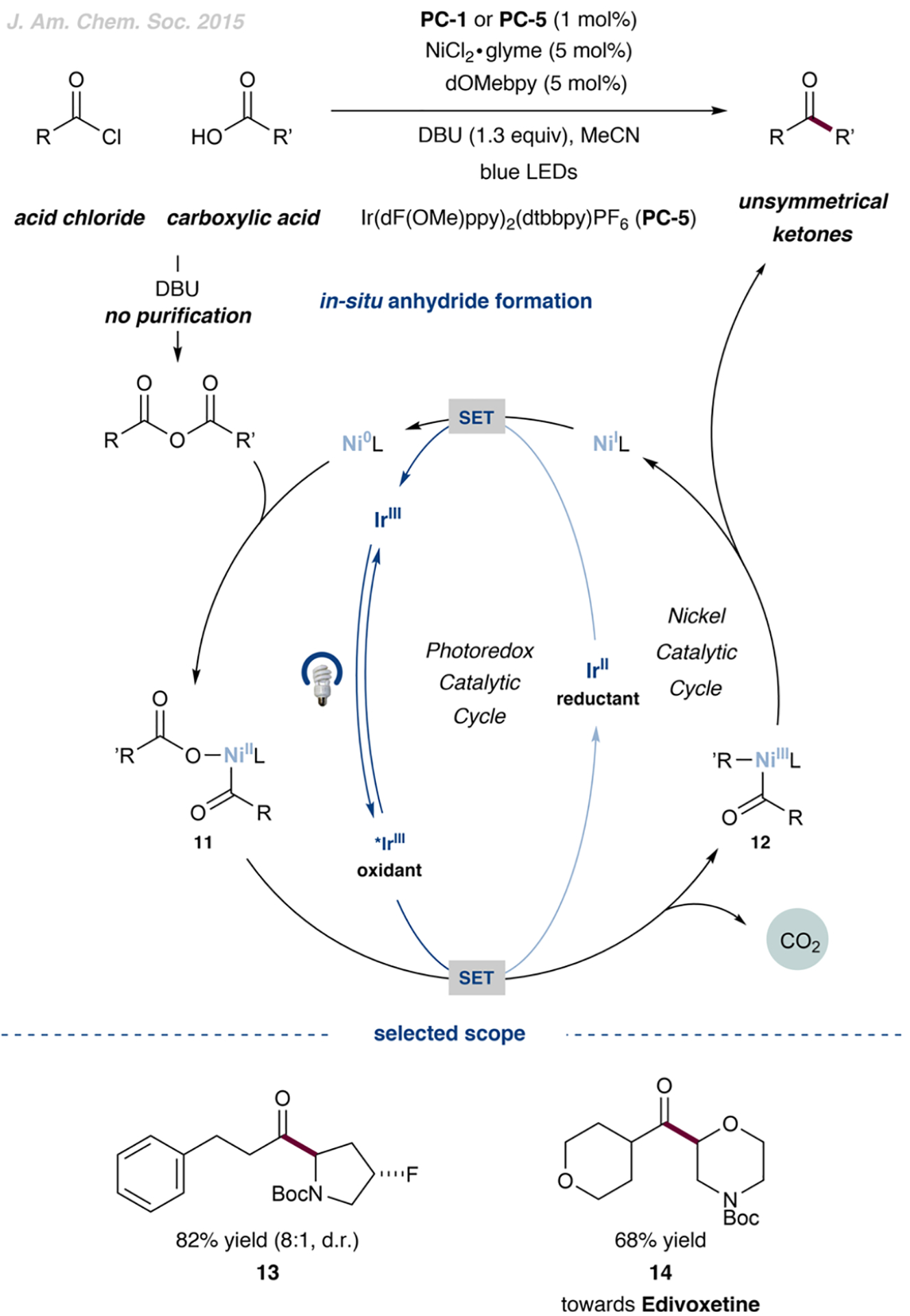
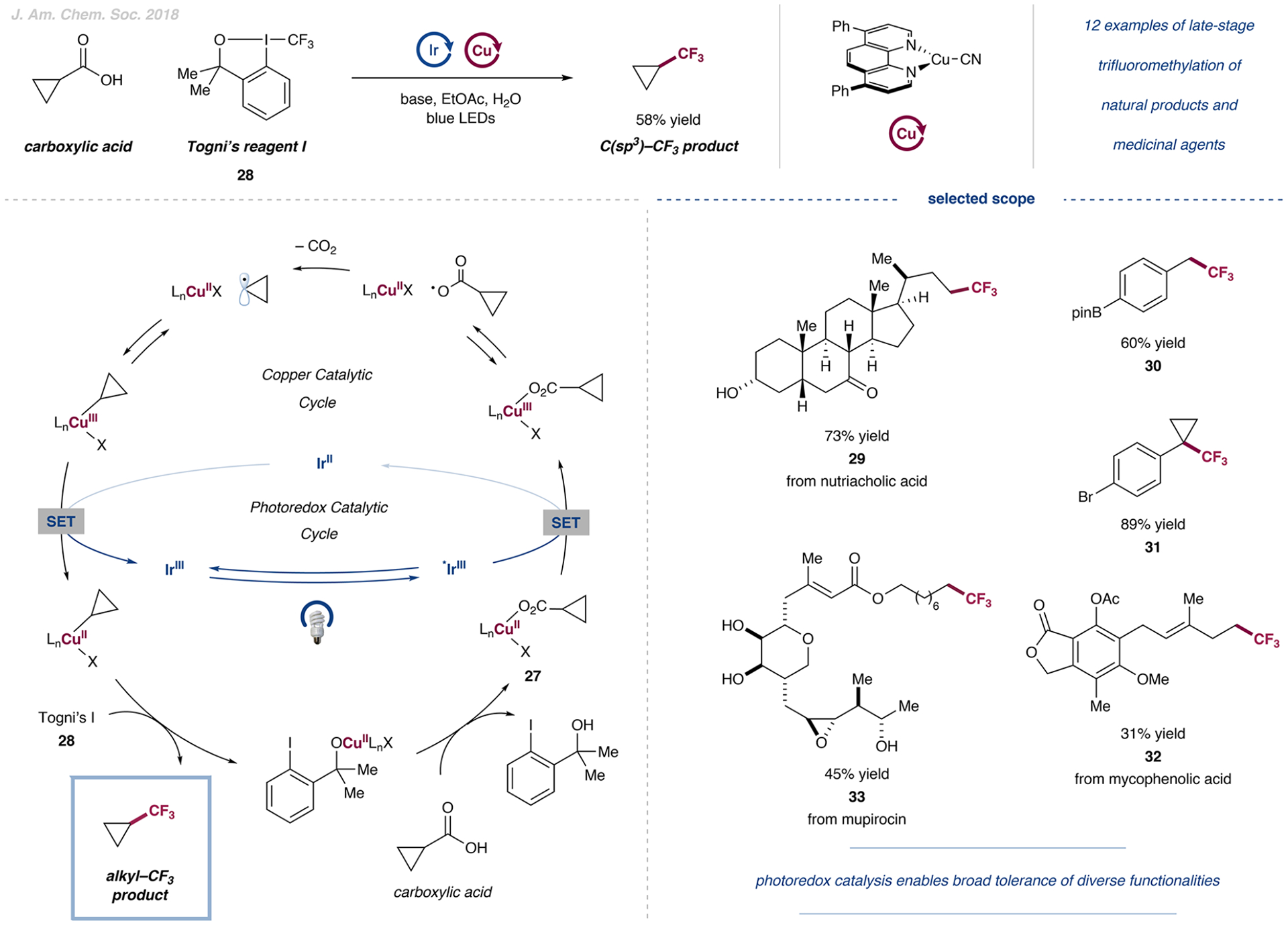
The development of palladium-catalyzed cross-coupling methods for the activation of C(sp2)-Br bonds facilitated access to arene-rich molecules, enabling a concomitant increase in the prevalence of this structural motif in drug molecules in recent decades. Today, there is a growing appreciation of the value of incorporating saturated C(sp3)-rich scaffolds into pharmaceutically active molecules as a means to achieve improved solubility and physiological stability, providing the impetus to develop new coupling strategies to access these challenging motifs in the most straightforward way possible. As an alternative to classical two-electron chemistry, redox chemistry can enable access to elusive transformations, most recently, by interfacing abundant first-row transition-metal catalysis with photoredox catalysis. As such, the functionalization of ubiquitous and versatile functional handles such as (aliphatic) carboxylic acids via metallaphotoredox catalysis has emerged as a valuable field of research over the past eight years.In this Account, we will outline recent progress in the development of methodologies that employ aliphatic and (hetero)aromatic carboxylic acids as adaptive functional groups. Whereas recent decarboxylative functionalization methodologies often necessitate preactivated aliphatic carboxylic acids in the form of redox-active esters or as ligands for hypervalent iodine reagents, methods that enable the direct use of the native carboxylic acid functionality are highly desired and have been accomplished through metallaphotoredox protocols. As such, we found that bench-stable aliphatic carboxylic acids can undergo diverse transformations, such as alkylation, arylation, amination, and trifluoromethylation, by leveraging metallaphotoredox catalysis with prevalent first-row transition metals such as nickel and copper. Likewise, abundant aryl carboxylic acids are now able to undergo halogenation and borylation, enabling new entry points for traditional, primarily palladium- or copper-catalyzed cross-coupling strategies. Given the breadth of the functional group tolerance of the employed reaction conditions, the late-stage functionalization of abundant carboxylic acids toward desired targets has become a standard tool in reaction design, enabling the synthesis of various diversified drug molecules. The rapid rise of this field has positively inspired pharmaceutical discovery and will be further accelerated by novel reaction development. The achievement of generality through reaction optimization campaigns allows for future breakthroughs that can render protocols more reliable and applicable for industry. This article is intended to highlight, in particular, (i) the employment of aliphatic and (hetero)aryl carboxylic acids as powerful late-stage adaptive functional handles in drug discovery and (ii) the need for the further development of still-elusive and selective transformations.We strongly believe that access to native functionalities such as carboxylic acids as adaptive handles will further inspire researchers across the world to investigate new methodologies for complex molecular targets.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
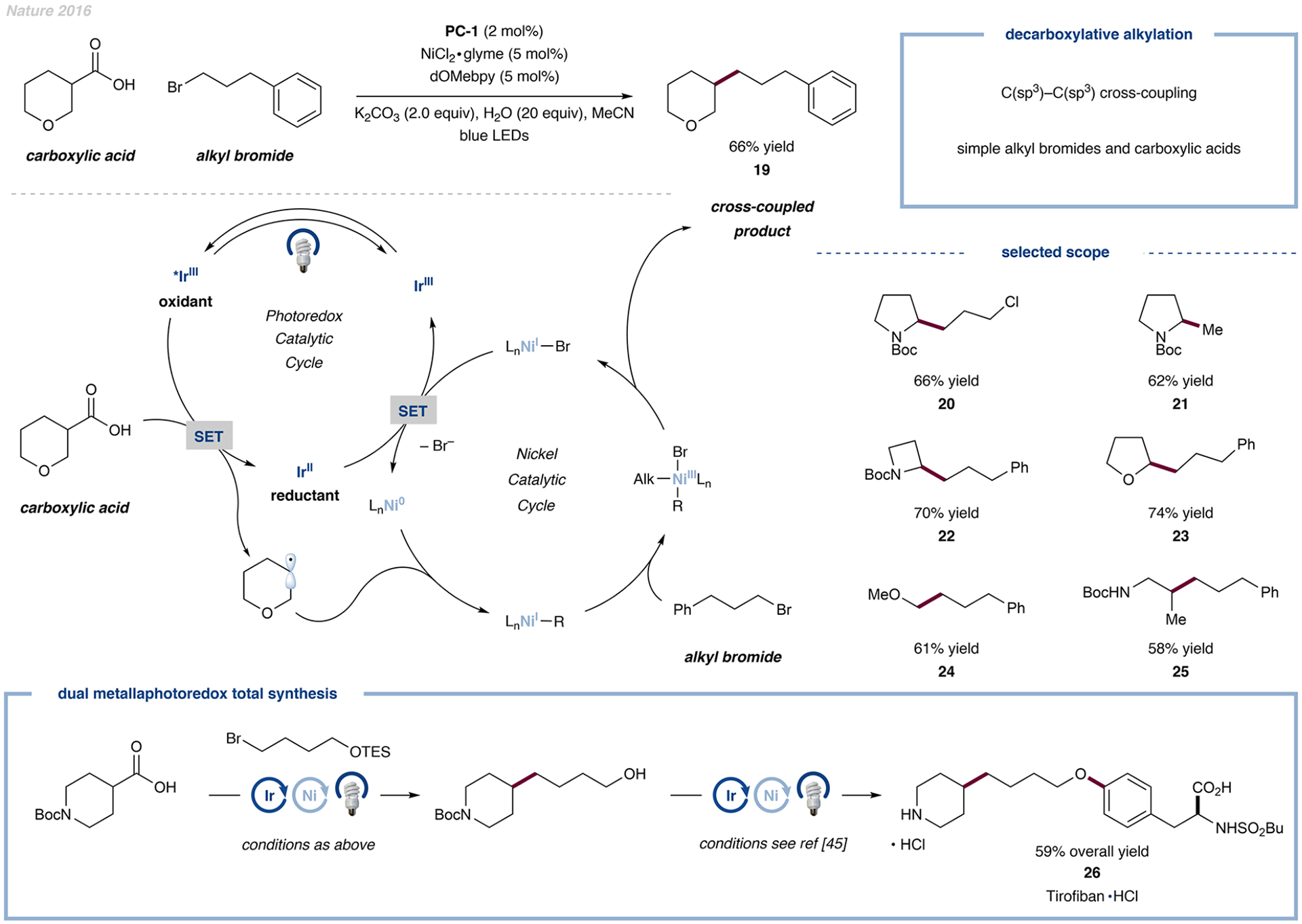
- Johnston CP; Smith RT; Allmendinger S; MacMillan DWC Metallaphotoredox-Catalysed Sp3–Sp3 Cross-Coupling of Carboxylic Acids with Alkyl Halides. Nature 2016, 536, 322–325. - PMC - PubMed
-
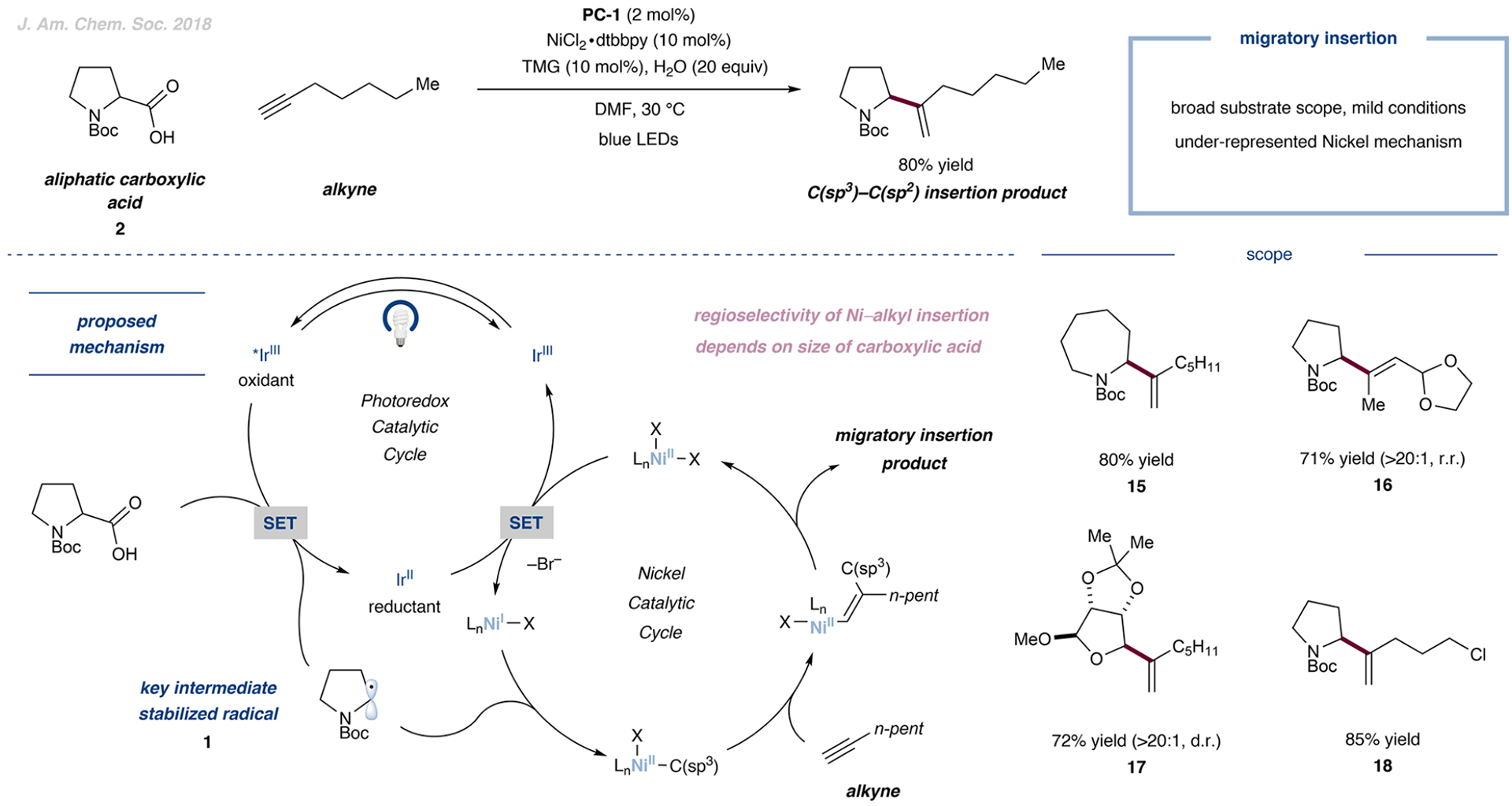
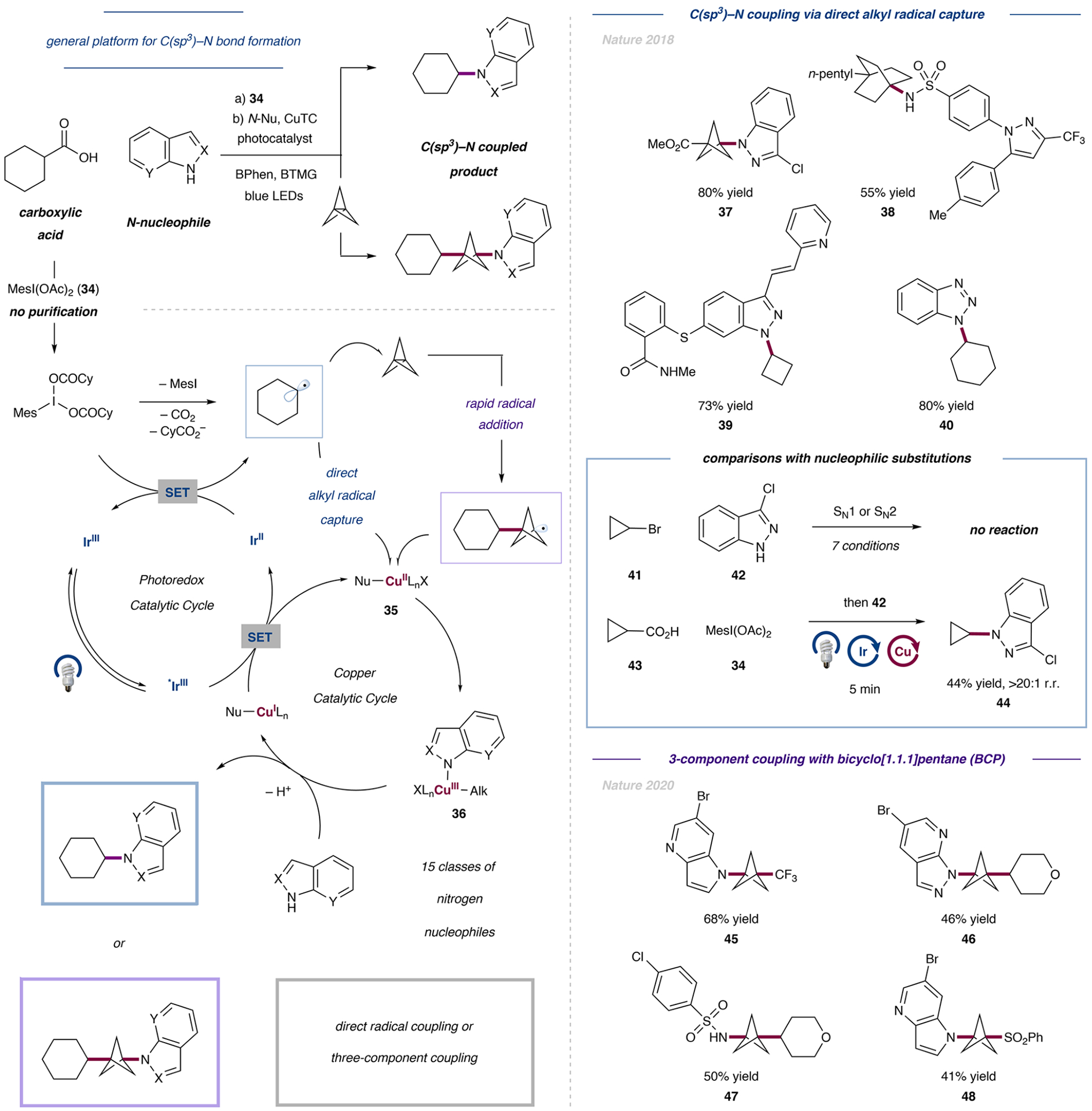
The transition-metal-catalyzed bond formation of two C(sp3) centers was previously unknown and was achieved by the independent activation of carboxylic acids and alkyl halides.
-
- Liang Y; Zhang X; MacMillan DWC Decarboxylative Sp3 C-N Coupling via Dual Copper and Photoredox Catalysis. Nature 2018, 559, 83–88. - PMC - PubMed
-
N-Alkylation usually proceeds from the alkyl bromide and requires harsh reaction conditions. Even more challenging is the introduction of secondary and tertiary alkyl derivatives, which became possible by the combination of copper catalysis and a straightforward decarboxylation strategy.
-
- Zhang X; Smith RT; Le C; McCarver SJ; Shireman BT; Carruthers NI; MacMillan DWC Copper-Mediated Synthesis of Drug-like Bicyclopentanes. Nature 2020, 580, 220–226. - PMC - PubMed
-
The pharmacologic relevance of BCP derivatives is well-appreciated, yet efficient and modular syntheses are scarce. This work describes the difunctionalization of BCP derivatives with diverse substituents.
-
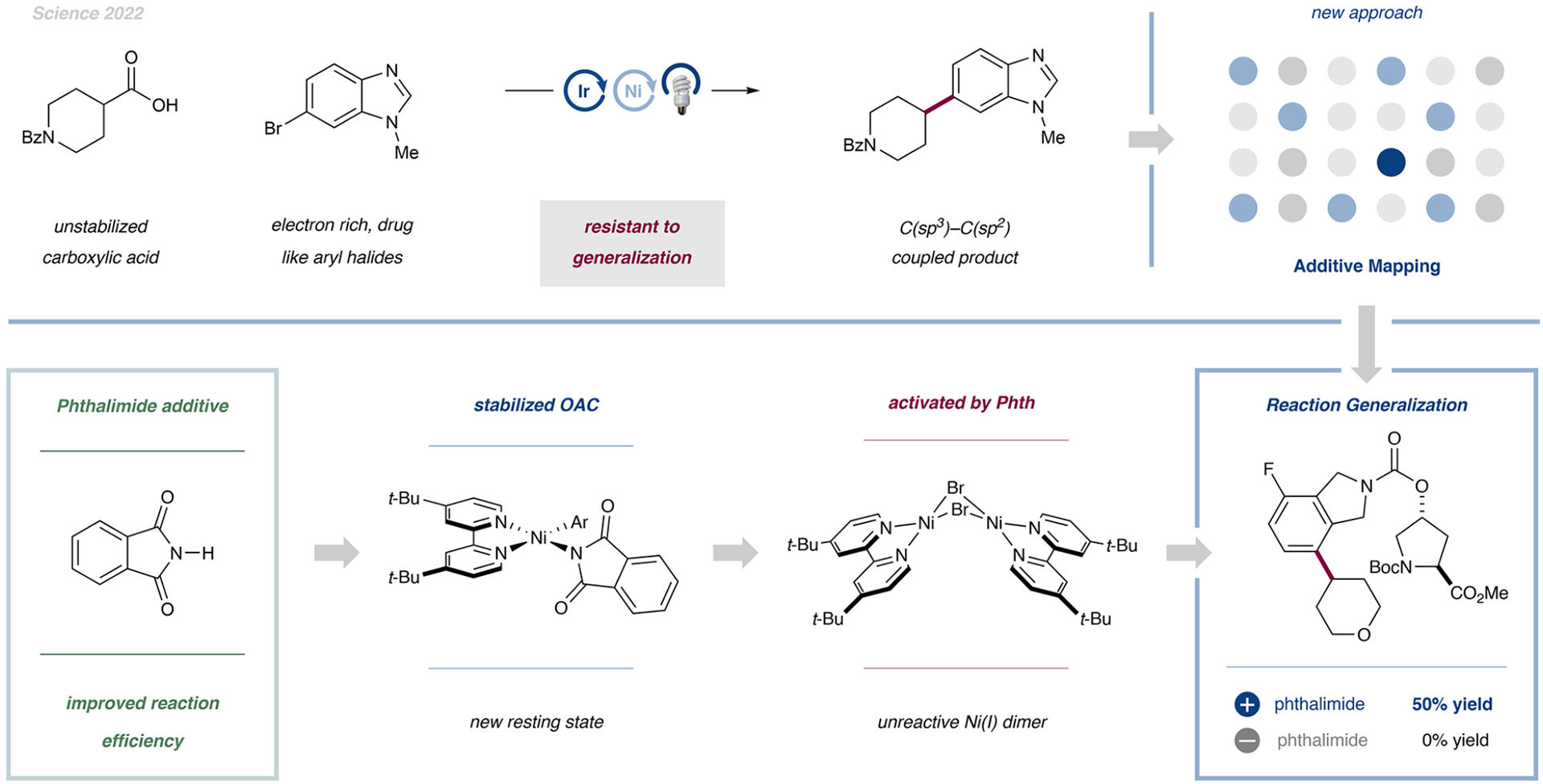
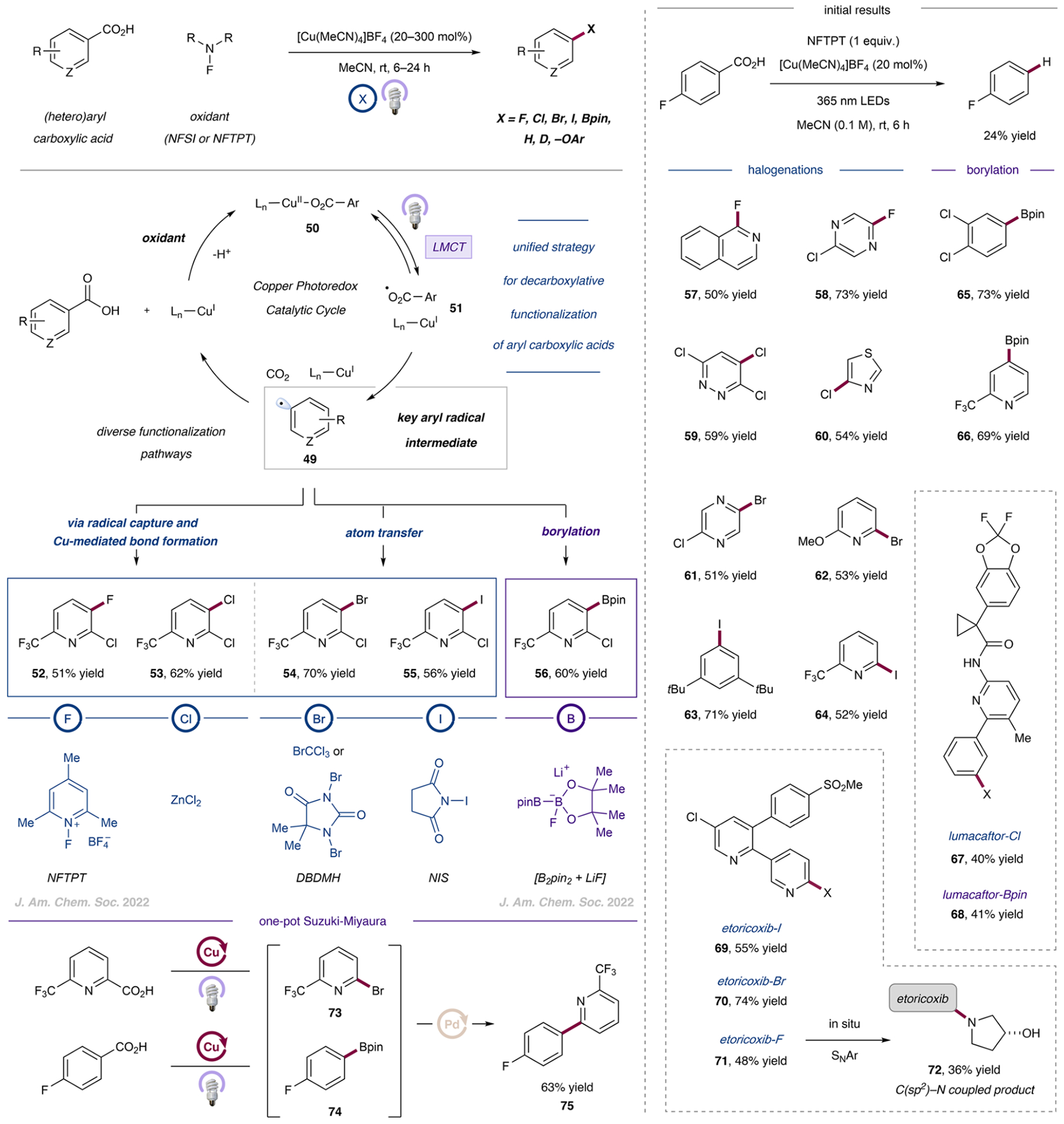
- Prieto Kullmer CN; Kautzky JA; Krska SW; Nowak T; Dreher SD; MacMillan DWC Accelerating Reaction Generality and Mechanistic Insight through Additive Mapping. Science 2022, 376, 532–539. - PMC - PubMed
-
A broader mechanistic understanding of the seminal metallaphotoredox decarboxylative arylation from 2014 led to the development of a broadly applicable procedure with improved performance. The identification of suitable additives, such as phthalimide, was the key to this generality.
-
- Li T; Huo L; Pulley C; Liu A Decarboxylation Mechanisms in Biological System. Bioorganic Chem 2012, 43, 2–14. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
