

Dysbiotic but nonpathogenic shift in the fecal mycobiota of patients with rheumatoid arthritis
- PMID: 36472468
- PMCID: PMC9728469
- DOI: 10.1080/19490976.2022.2149020
Dysbiotic but nonpathogenic shift in the fecal mycobiota of patients with rheumatoid arthritis
Abstract
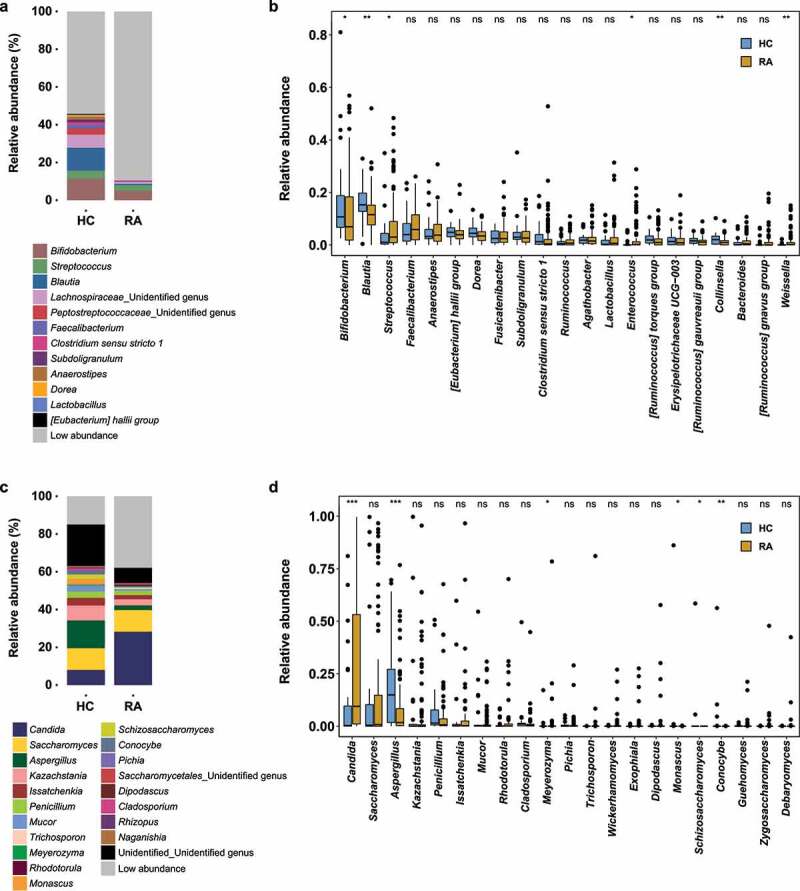
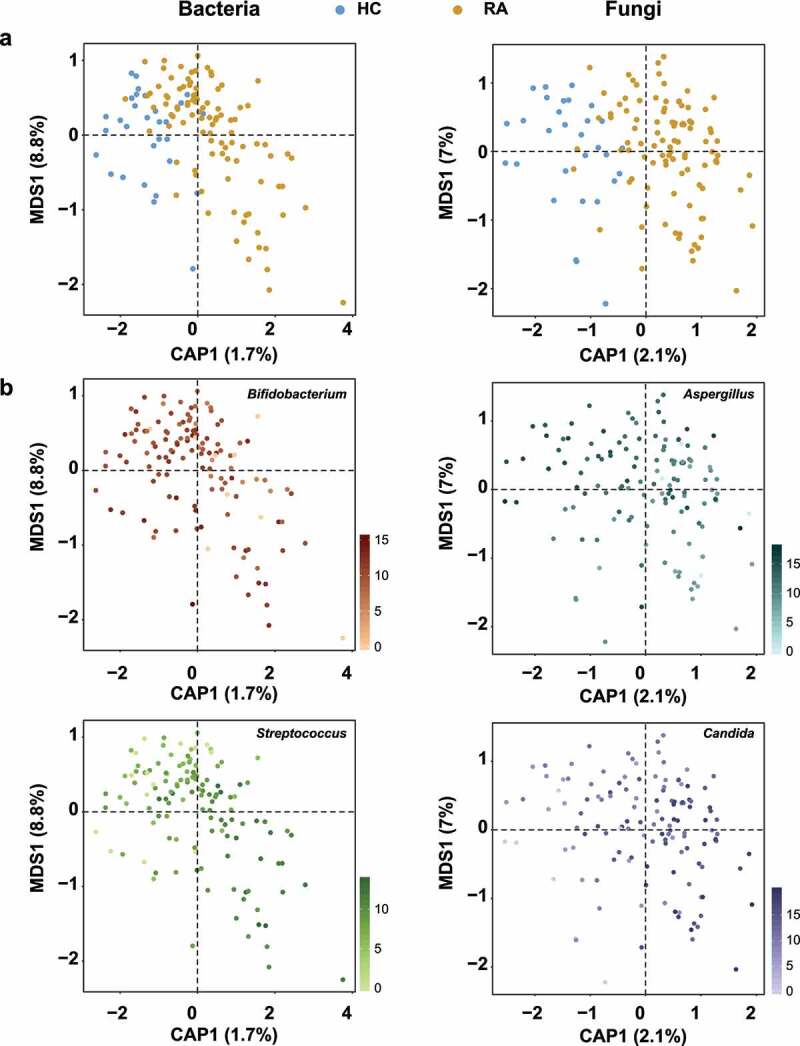
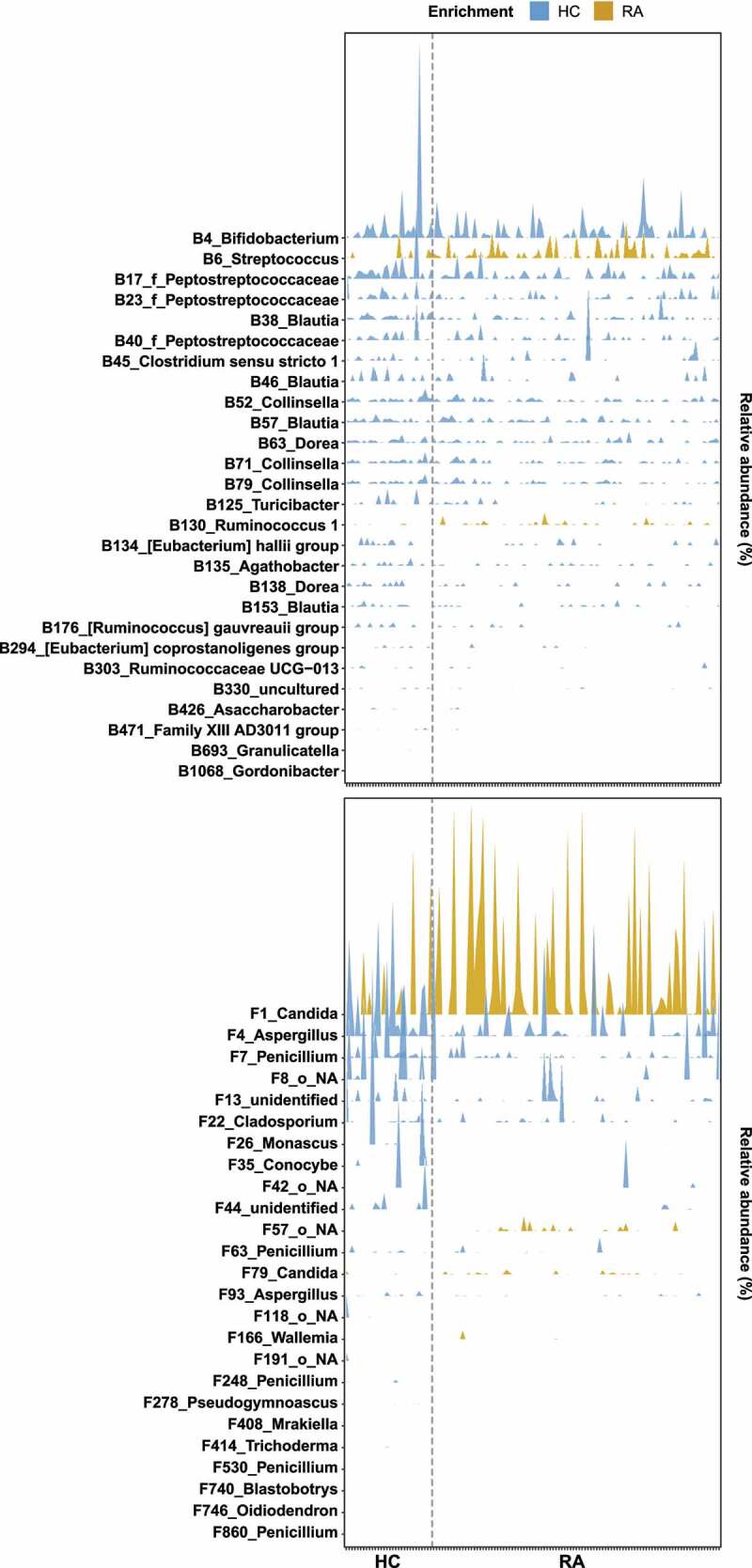
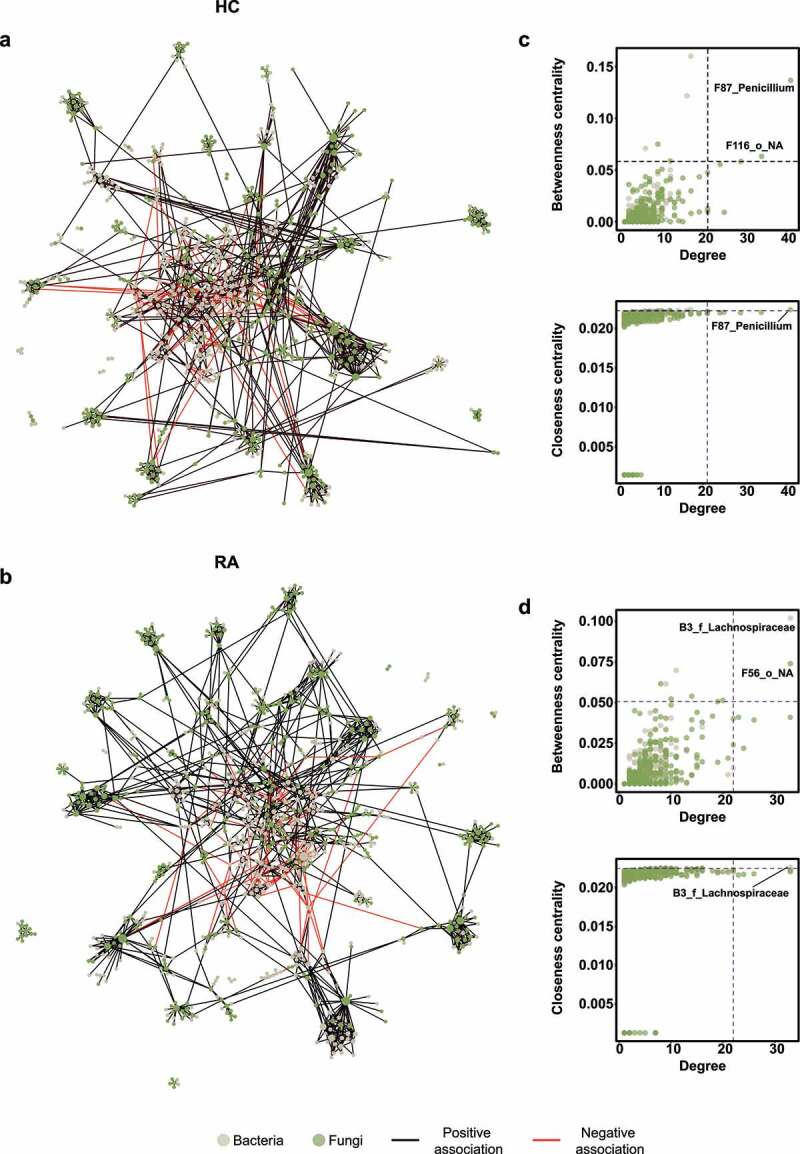
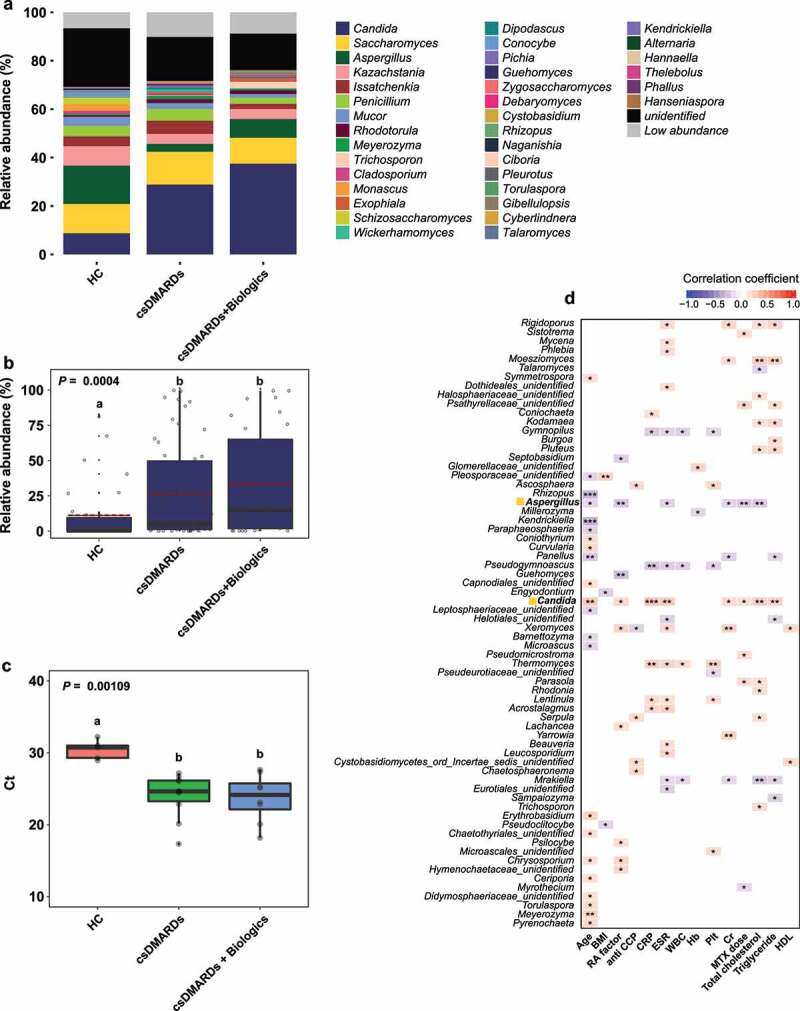
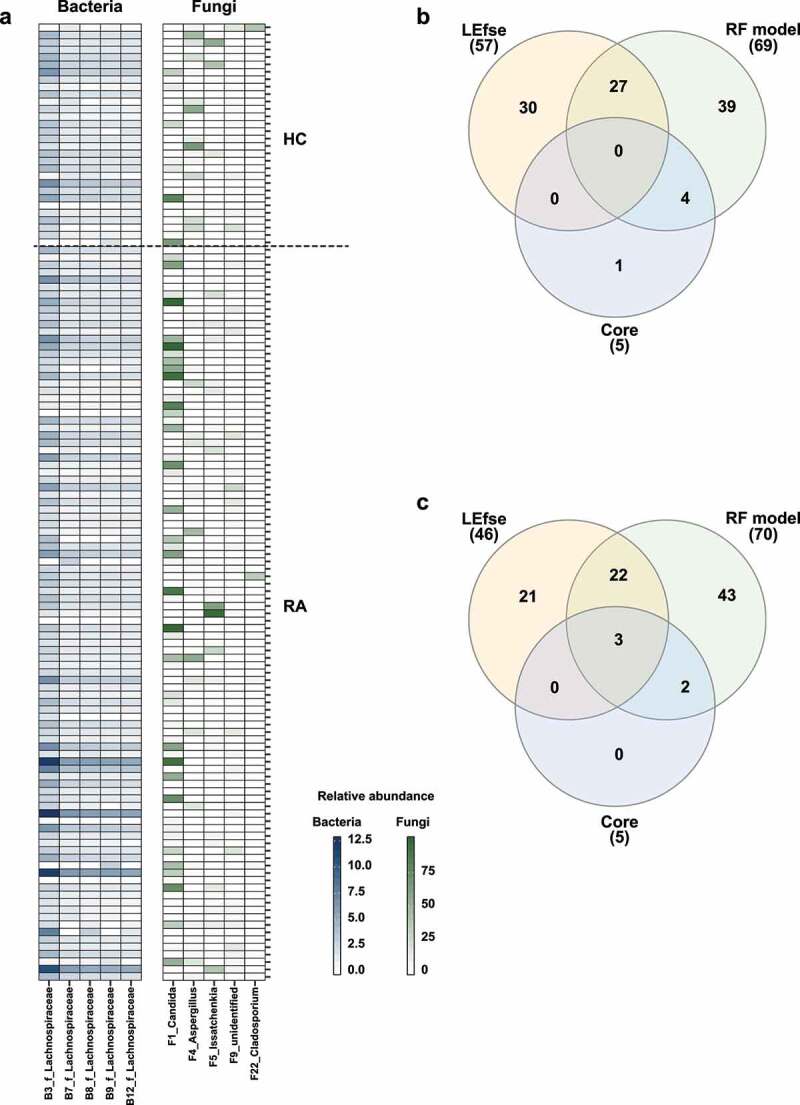
Rheumatoid arthritis (RA) is closely associated with the oral and gut microbiomes. Fungal cell wall components initiate inflammatory arthritis in mouse models. However, little is known regarding the role of the fungal community in the pathogenesis of RA. To evaluate the association between RA and the gut microbiome, investigations of bacterial and fungal communities in patients with RA are necessary. Therefore, we investigated the compositions and associations of fecal bacterial and fungal communities in 30 healthy controls and 99 patients with RA. The relative abundances of Bifidobacterium and Blautia decreased, whereas the relative abundance of Streptococcus increased, in patients with RA. The relative abundance of Candida in the fecal fungal community was higher in patients with RA than in healthy controls, while the relative abundance of Aspergillus was higher in healthy controls than in patients with RA. Candida species-specific gene amplification showed that C. albicans was the most abundant species of Candida. Ordination analysis and random forest classification models supported the findings of structural changes in bacterial and fungal communities. Aspergillus was the core fecal fungal genus in healthy controls, although Saccharomyces spp. are typically predominant in Western cohorts. In addition, bacterial-fungal association analyses showed that the hub node had shifted from fungi to bacteria in patients with RA. The finding of fungal dysbiosis in patients with RA suggests that fungi play critical roles in the fecal microbial communities and pathogenesis of RA.
Keywords: Aspergillus; Candida; Fecal microbiota; dysbiosis; rheumatoid arthritis.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Klareskog L, Catrina AI, Paget S. Rheumatoid arthritis. Lancet. 2009;373:659–672. - PubMed
-
- Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, Kavanaugh A, McInnes IB, Solomon DH, Strand V, et al. Rheumatoid arthritis. Nat Rev Dis Primers. 2018;4:18001. - PubMed
-
- Tsuchiya Y, Takayanagi N, Sugiura H, Miyahara Y, Tokunaga D, Kawabata Y, Sugita Y. Lung diseases directly associated with rheumatoid arthritis and their relationship to outcome. Eur Respir J. 2011;37:1411–1417. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous