

Spatial transcriptomic characterization of COVID-19 pneumonitis identifies immune circuits related to tissue injury
- PMID: 36472908
- PMCID: PMC9977306
- DOI: 10.1172/jci.insight.157837
Spatial transcriptomic characterization of COVID-19 pneumonitis identifies immune circuits related to tissue injury
Abstract
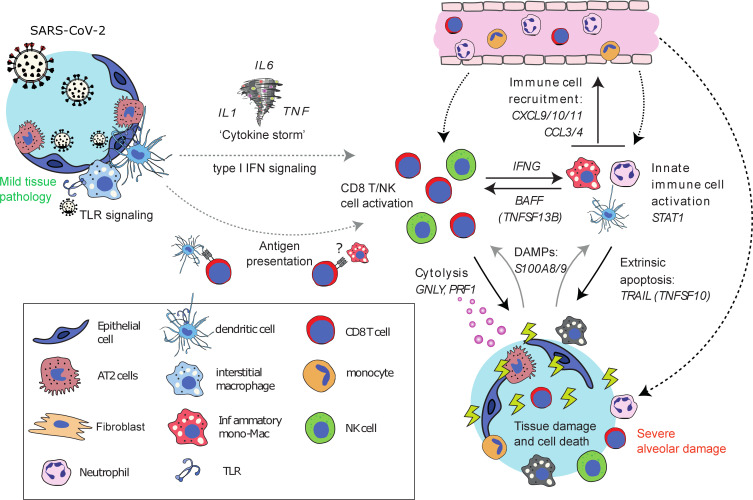
Severe lung damage resulting from COVID-19 involves complex interactions between diverse populations of immune and stromal cells. In this study, we used a spatial transcriptomics approach to delineate the cells, pathways, and genes present across the spectrum of histopathological damage in COVID-19-affected lung tissue. We applied correlation network-based approaches to deconvolve gene expression data from 46 areas of interest covering more than 62,000 cells within well-preserved lung samples from 3 patients. Despite substantial interpatient heterogeneity, we discovered evidence for a common immune-cell signaling circuit in areas of severe tissue that involves crosstalk between cytotoxic lymphocytes and pro-inflammatory macrophages. Expression of IFNG by cytotoxic lymphocytes was associated with induction of chemokines, including CXCL9, CXCL10, and CXCL11, which are known to promote the recruitment of CXCR3+ immune cells. The TNF superfamily members BAFF (TNFSF13B) and TRAIL (TNFSF10) were consistently upregulated in the areas with severe tissue damage. We used published spatial and single-cell SARS-CoV-2 data sets to validate our findings in the lung tissue from additional cohorts of patients with COVID-19. The resulting model of severe COVID-19 immune-mediated tissue pathology may inform future therapeutic strategies.
Keywords: COVID-19; Cellular immune response; Chemokines; Inflammation; Molecular pathology.
Figures





References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
