

Performance assessment and economic analysis of a human Liver-Chip for predictive toxicology
- PMID: 36473994
- PMCID: PMC9727064
- DOI: 10.1038/s43856-022-00209-1
Performance assessment and economic analysis of a human Liver-Chip for predictive toxicology
Erratum in
-
Author Correction: Performance assessment and economic analysis of a human Liver-Chip for predictive toxicology.Commun Med (Lond). 2023 Jan 12;3(1):7. doi: 10.1038/s43856-023-00235-7. Commun Med (Lond). 2023. PMID: 36635369 Free PMC article. No abstract available.
-
Author Correction: Performance assessment and economic analysis of a human Liver-Chip for predictive toxicology.Commun Med (Lond). 2023 Feb 2;3(1):16. doi: 10.1038/s43856-023-00249-1. Commun Med (Lond). 2023. PMID: 36732600 Free PMC article. No abstract available.
Abstract
Background: Conventional preclinical models often miss drug toxicities, meaning the harm these drugs pose to humans is only realized in clinical trials or when they make it to market. This has caused the pharmaceutical industry to waste considerable time and resources developing drugs destined to fail. Organ-on-a-Chip technology has the potential improve success in drug development pipelines, as it can recapitulate organ-level pathophysiology and clinical responses; however, systematic and quantitative evaluations of Organ-Chips' predictive value have not yet been reported.
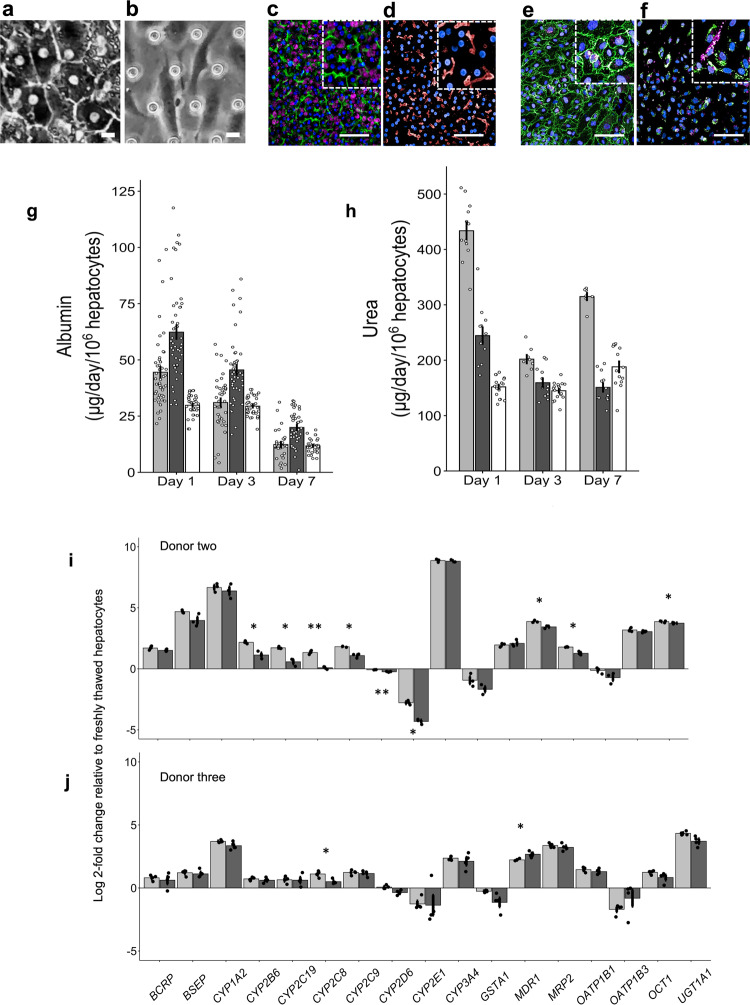
Methods: 870 Liver-Chips were analyzed to determine their ability to predict drug-induced liver injury caused by small molecules identified as benchmarks by the Innovation and Quality consortium, who has published guidelines defining criteria for qualifying preclinical models. An economic analysis was also performed to measure the value Liver-Chips could offer if they were broadly adopted in supporting toxicity-related decisions as part of preclinical development workflows.
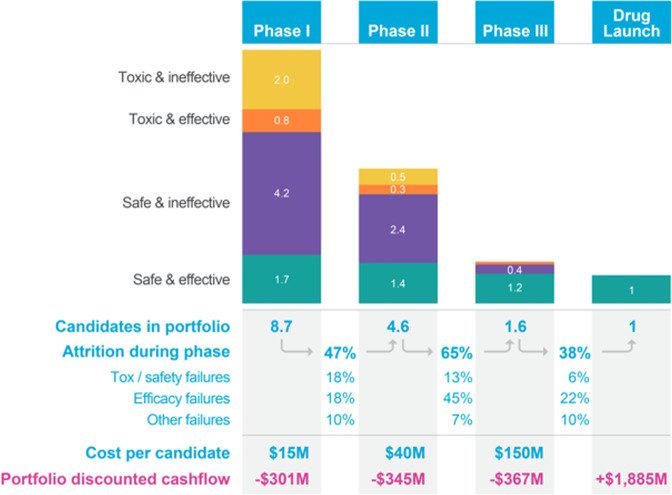
Results: Here, we show that the Liver-Chip met the qualification guidelines across a blinded set of 27 known hepatotoxic and non-toxic drugs with a sensitivity of 87% and a specificity of 100%. We also show that this level of performance could generate over $3 billion annually for the pharmaceutical industry through increased small-molecule R&D productivity.
Conclusions: The results of this study show how incorporating predictive Organ-Chips into drug development workflows could substantially improve drug discovery and development, allowing manufacturers to bring safer, more effective medicines to market in less time and at lower costs.
Plain language summary
Drug development is lengthy and costly, as it relies on laboratory models that fail to predict human reactions to potential drugs. Because of this, toxic drugs sometimes go on to harm humans when they reach clinical trials or once they are in the marketplace. Organ-on-a-Chip technology involves growing cells on small devices to mimic organs of the body, such as the liver. Organ-Chips could potentially help identify toxicities earlier, but there is limited research into how well they predict these effects compared to conventional models. In this study, we analyzed 870 Liver-Chips to determine how well they predict drug-induced liver injury, a common cause of drug failure, and found that Liver-Chips outperformed conventional models. These results suggest that widespread acceptance of Organ-Chips could decrease drug attrition, help minimize harm to patients, and generate billions in revenue for the pharmaceutical industry.
© 2022. The Author(s).
Conflict of interest statement
The authors declare the following competing interests: L.E., D.L., D.V.M., J.D.S., A.A., S.A.B., J.T.C., C.V.C., A.R.H., J.J., S.J., S.R.J., J.F.K.S., M.M.K., M.K., K.K.M., M.E.Q., A.C.R., W.T.S., M.W., G.K., V.J.K., C.Y.L., C. L., J.S.R., D.R.T., J.V., and K.-J.J. are employees or former employees of Emulate Inc. and may hold equity; D.E.I. is a founder, board member, SAB chair, and equity holder in Emulate Inc. J.W.S. is a shareholder and director of JW Scannell Analytics LTD and received payment from Emulate Inc. for contributing to this work. O.I. and P.K.M. have no competing interests to share.
Figures



References
-
- Khanna I. Drug discovery in pharmaceutical industry: productivity challenges and trends. Drug Discov. Today. 2012;17:1088–1102. - PubMed
-
- Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004;3:711–716. - PubMed
-
- Peck RW, Lendrem DW, Grant I, Lendrem BC, Isaacs JD. Why is it hard to terminate failing projects in pharmaceutical R&D? Nat. Rev. Drug Discov. 2015;14:663–664. - PubMed
-
- Paul SM, et al. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010;9:203–214. - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
