

Mitochondrial biology and dysfunction in secondary mitochondrial disease
- PMID: 36475414
- PMCID: PMC9727669
- DOI: 10.1098/rsob.220274
Mitochondrial biology and dysfunction in secondary mitochondrial disease
Abstract
Mitochondrial diseases are a broad, genetically heterogeneous class of metabolic disorders characterized by deficits in oxidative phosphorylation (OXPHOS). Primary mitochondrial disease (PMD) defines pathologies resulting from mutation of mitochondrial DNA (mtDNA) or nuclear genes affecting either mtDNA expression or the biogenesis and function of the respiratory chain. Secondary mitochondrial disease (SMD) arises due to mutation of nuclear-encoded genes independent of, or indirectly influencing OXPHOS assembly and operation. Despite instances of novel SMD increasing year-on-year, PMD is much more widely discussed in the literature. Indeed, since the implementation of next generation sequencing (NGS) techniques in 2010, many novel mitochondrial disease genes have been identified, approximately half of which are linked to SMD. This review will consolidate existing knowledge of SMDs and outline discrete categories within which to better understand the diversity of SMD phenotypes. By providing context to the biochemical and molecular pathways perturbed in SMD, we hope to further demonstrate the intricacies of SMD pathologies outside of their indirect contribution to mitochondrial energy generation.
Keywords: mitochondria; mitochondrial protein import; mitochondrial quality control; secondary mitochondrial disease.
Conflict of interest statement
We declare we have no competing interests.
Figures
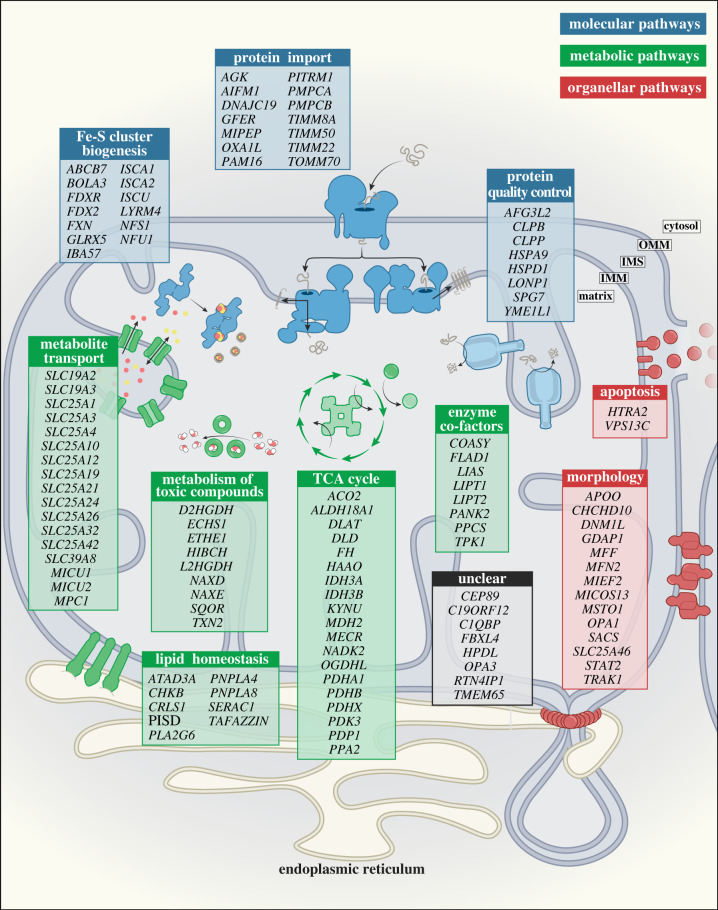
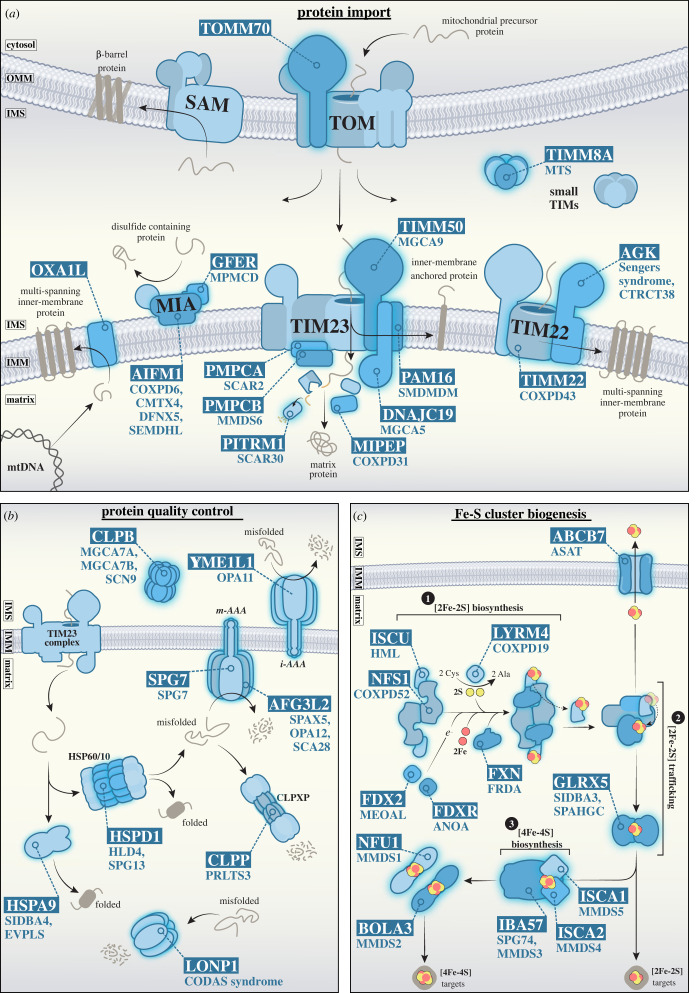
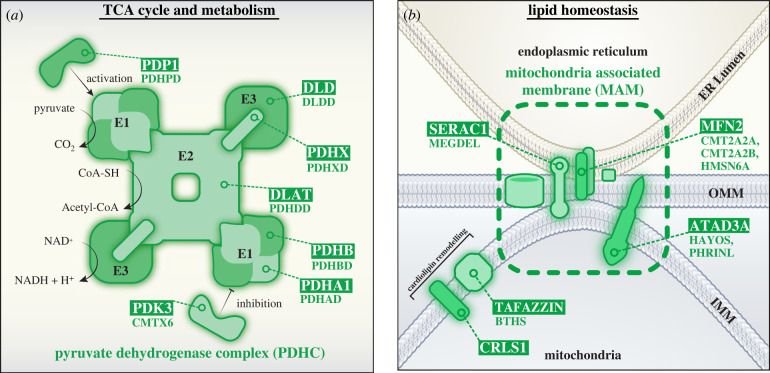
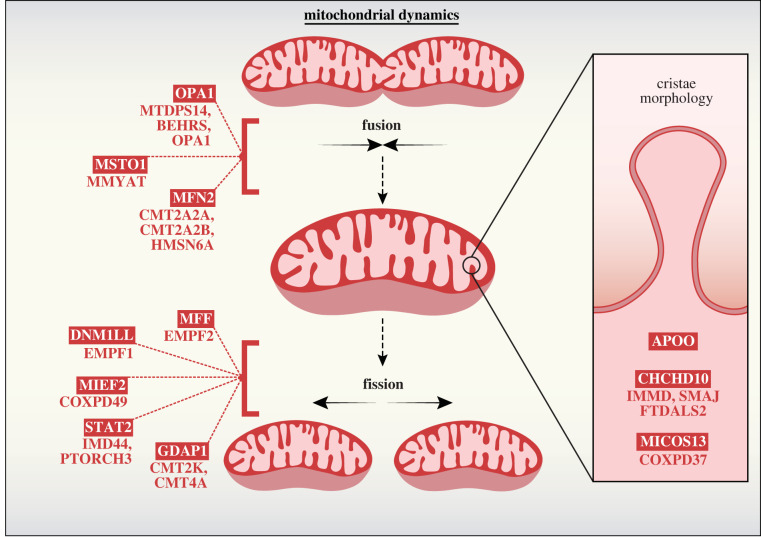
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
